
Abstract
MicroRNA (miRNA) has emerge as a vital regulator in the pathogenesis of intervertebral disc degeneration (IDD). However, miR-106b-5p expression in the human nucleus pulposus (NP) and potential mechanisms remain to be elucidated. In this study, the aim was to verify the potential therapeutic mechanisms of miR-106b-5p for IDD. Key miRNAs were screened for in degenerative and normal human intervertebral disc samples. qRT-PCR and fluorescence in situ hybridization (FISH) were used to verify the miR-106b-5p differential expression. The targeting link between miR-106b-5p and Sirtuin 2 (SIRT2) was identified using the luciferase reporter assay and bioinformatics. Flow cytometry, EdU method, and cell scratching were all performed to determine the NP cell function and IDD models were constructed for in vivo experiments. SIRT2, MMP13, ADAMTS5, Col II, Aggrecan, Ras, ERK1/2, and p-ERK1/2 protein levels were assayed by western blotting. Overexpression of miR-106b-5p in NP cells decreased cell growth, induced apoptosis, hindered extracellular matrix formation, and increased the expression of matrix-degrading enzymes through the SIRT2/MAPK/ERK signaling pathway. Importantly, intradiscal delivery of antagomiR-106b-5p significantly attenuated IDD development. Our findings demonstrate that targeting miR-106b-5p in intervertebral disc has therapeutic effects on IDD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Low back pain (LBP) is a common manifestation of spinal musculoskeletal disorders [1, 2]. Approximately 84% of people will encounter LBP during their lifetime, making LBP a social burden [3, 4]. Intervertebral disc degeneration (IDD) is a frequent contributor to lumbar spine disease [5]. Current treatments for IDD, including physical therapy, medications and spinal fusion, only focus on alleviating or managing pain. None of these approaches are precise treatments that target the pathogenesis to reverse the course of IDD [6]. Thus, it is particularly important to elucidate the pivotal molecular mechanisms of IDD and seek innovative therapeutic strategies. Abnormal expression of miRNAs has been implicated in the pathogenesis of IDD, and miRNA-based therapeutic approaches emerge as a promising treatment strategy [7,8,9].
MicroRNA (miRNA) is an endogenous non-coding RNA that negatively regulates the stability and/or inhibits the translation of messenger RNAs (mRNAs) by binding to specific sequences within the target mRNA [10]. Although miRNAs are non-coding, they are involved in the regulation of approximately 30% of human proteins, as well as the regulation of cellular proliferation, apoptosis, and differentiation of a wide range of cells [11]. Abnormal expression of miRNA has been found in a number of human musculoskeletal disorders [12, 13]. Recent studies have identified an important role for miRNAs in the progress of IDD [14,15,16], including cell proliferation, apoptosis, extracellular matrix (ECM) degradation, senescence, and inflammation. Correspondingly, there is support for selection of miRNAs as potential therapeutic targets for IDD treatment [17]. miR-106b-5p is a member of the miR-106b-25 cluster and is reported to be involved in the regulation of some musculoskeletal degenerative diseases [18, 19]. However, the mechanism of miR-106b-5p regulation in IDD is still yet to be studied.
Here, this study aims to elucidate the mechanism through which miR-106b-5p regulates nucleus pulposus (NP) cell function, and explores the effectiveness of antagomiR-106b-5p in balancing the anabolic and catabolic metabolism of NP in vivo. Overall, this may provide the necessary experimental basis for the development of effective therapeutic approaches based on intradiscal injection of miRNA drugs.
Materials and methods
Patient samples
NP samples were obtained from a total of 212 patients diagnosed with degenerative disc disease (mean age, 55.6 ± 9.4 years). Indications for surgery include the following: (1) ineffectual conservative therapy; (2) worsening neurological impairments, such as progressive dyskinesia, cauda equina syndrome, (3) severe intervertebral disc degeneration (Pfirrmann’s grade IV or V). Otherwise, the following conditions should first be excluded: (1) lumbar infection or (2) ankylosing spondylitis, or (3) previous lumbar procedures. Eighty NP samples were collected for the control group, which included patients who underwent spinal procedures for fresh vertebral burst fractures (mean age, 54.3 ± 7.4 years). All the included patients had lumbosacral magnetic resonance imaging, as part of their routine preoperative evaluation. T2-weighted images were used as the Pfirrmann classification for grading the degree of disc degeneration [20]. The subjects with Pfirrmann’s grade I were included in control group. The ethics committee of The First Affiliated Hospital of Guangxi Medical University approved this study (2018-KY-NSFC-025). All patients enrolled in the study signed informed consent, and in cases where patients were unable to sign themselves, their designated family member(s) signed by proxy prior to the collection of the surgically removed disc tissues.
Surgically establishment of IDD model and miR-106b-5p therapeutic approach
Annulus fibrosus (AF) needle puncture was used to establish the IDD model as previously described [21, 22]. Mice caudal vertebrae were chosen to build the model since the caudal disc of mice is thought to be comparable to human discs [23, 24]. The mice were subjected to general anesthesia via inhaled isoflurane. Following the complete exposure of the coccygeal discs (Co6/Co7), a needle attached to a 31G syringe was injected into the NP center, parallel to the endplates, and rotated 180° for 10 s.
A total of 20 C57BL/6 mice (12 weeks old, 23 ± 2 g) were obtained from The Guangxi Medical University Animal Center for the miR-106b-5p rescue investigation. Twenty mice were randomly allocated into four separate groups: the agomiR NC group, the agomiR-106b-5p group, the antagomiR NC group, and the antagomiR- 106b-5p group. Following IDD surgery, the mice given were injected with 10µL solutions of agomiR-106b-5p, antagomiR-106b-5p or their respective negative controls on 1 day, 1 week, and 2 weeks respectively. The IDD mice were euthanized after the 12th week of intervention, and their disc tissues were taken for histological staining and immunohistochemistry studies. The ethics committee of The First Affiliated Hospital of Guangxi Medical University approved all animal testing protocols (2018-KY-NSFC-025).
Human NP cells isolation and culture
All protocols for the extraction of NP cells were followed exactly [25]. Tissues were washed three times in PBS, cut into 2 mm3 pieces, digested with 0.25 trypsin (Thermo Fisher Scientific, MA, USA) and 2% type II collagenase (Invitrogen, CA, USA), and finally kept in PBS for three hours at 37℃. The cells were then filtered using a 200-mesh filter. Primary human NP cells were inoculated in a 25-cm culture dish and cultured with DMEM (Gibco, NY, USA) containing 10% FBS (Biological Industries, Israel) and 1% penicillin/streptomycin (Biological Industries, Israel) in a 5% CO2 incubator at 37 °C. Change the culture medium every 2 days. When the cell density reaches 80%, the cells are passaged at 1:2. No obvious differences in cell morphology existed between the two generations. The well-grown cells (second generation) were taken to subsequent experiments.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated and extracted from collected NP tissues using Trizol reagent (Takara, Aichi Ken, Japan). Afterward, reverse transcription was performed using the Prime Script miRNA cDNA Synthesis Kit (Takara, Aichi Ken, Japan). Differential expression of SIRT2 and miR-106b-5p was analyzed by 7500 real-time PCR system (Thermo Fisher Scientific, MA, USA) and TB Green Premix Ex Taq (Takara, Aichi Ken, Japan). The miRNA and mRNA levels were then compared to U6 and GAPDH, respectively, as standard controls. The 2−ΔΔCt was implemented to calculate the relative changes. The primer sequences used in this study are displayed in Supplementary Table 1.
Cell transfection
The miR-106b-5p mimics, miR-106b-5p mimics control, miR-106b-5p inhibitor and miR-106b-5p inhibitor control were provided by Genechem Company (Shanghai, China). The NP cells were seeded in a 6-well plate, and transfected at 20–30% cell density using Lipofectamine 3000 (Invitrogen, CA, USA). Following 72 h of transfection, the related analysis was performed. The SIRT2 CDS sequence was synthesized and sub-cloned into the pCDNA3.1 (Invitrogen) vector. Overexpression of SIRT2 was achieved via pCDNA3.1-SIRT2 transfection. Harvest approximately 1 × 105 cells and inoculate them in 6-well cell culture plates. The SIRT2 expression plasmids were transfected using Lipofectamine PLUS™ (Invitrogen, CA, USA) reagent when the cell growth density reached approximately 80%. The experiments were divided into control, miR-106b-5p, miR-106b-5p + SIRT2 and SIRT2 groups. Cell lysates were collected 48 h after transfection for the next step of the experiment.
miRNA microarrays
Initially, total RNA (50 ng) was extracted from the three IDD NP samples and the three control NP samples using the Trizol reagent. The miRNA fraction of the total RNA was next purified utilizing a miRNA kit (Thermo Fisher Scientific, MA, USA). Finally, the Affymetrix4.0 miRNA array was used to conduct miRNA microarray investigations (Thermo Fisher Scientific, MA, USA). The Gene Spring GXv12.1 software suite was applied to process the results consistently (Agilent Technologies). Fold change filtering was used to discover changes in miRNA expression between the two samples. Gene Cluster 3.0 software from Stanford University was used for hierarchical clustering analysis. The KEGG Homology Annotation System was used to perform functional enrichment analysis (KOBAS) [26]. GO analysis was also performed for differentially expressed genes.
miR-106b-5p target prediction and luciferase reporter assay
Using the miRbase database, we queried the specific microRNA basic information. Bioinformatics online analytical tools were taken to predict the target genes. Gene Ontology (GO) analysis was performed using the David Bioinformatics program to predict the effect of targeted mRNA on the disc. Targetscan was used to predict the 3’ non-coding region of the SIRT2 gene binding sites to the miR-106b-5p. The mutant (MUT) and wild-type (WT) 3’ UTRs of the SIRT2 gene fragments were created and cloned from human genomic DNA and put into the psiCHECK-2 vector’s downstream single restriction enzyme sites of the Renilla luciferase stop codon (Promega, WI, USA). Using Lipofectamine 2000, the NP cells were transfected with a reporter plasmid containing the WT or MUT 3’ UTR and miR-106b-5p mimic, alongside the miR-106b-5p inhibitor as a control. After 24 h, the luciferase activities were assessed using the GENios Pro (Promega, WI, USA).
Western blotting
Western blotting was performed by standard protocols. The total proteins in the NP cells were extracted with RIPA buffer supplemented with a protease inhibitor cocktail. The subsequent protein concentration and purity were assessed using a BCA kit (Beyotime, Shanghai, China). The proteins were loaded onto an SDS-PAGE gel before being transferred to a PVDF membrane (Solarbio, Beijing, China). The membrane was then blocked for 1 h with 5% nonfat dry milk before being treated overnight with primary antibodies at 4 °C. Afterward, the membranes were washed with TBST and incubated with the appropriate secondary antibodies for 2 h. The main antibodies were used: anti-Col II (Abcam: ab34712), anti-Aggrecan (Abcam: ab36861), anti-MMP13 (Abcam: ab219620), anti-ADAMTS5 (Abcam: ab219620), anti-ERK1/2 (Abcam: ab184699), anti-p-ERK1/2 (Abcam: ab184699), anti-Ras (Abcam: ab108602), anti-SIRT2 (Abeam: ab134171), anti-β-Actin (Abcam: ab179467), anti-GAPDH (Abcam: ab9485), which were normalized by blotting the same membrane with an antibody against GAPDH and β-Actin. ECL Detection Reagent (Solarbio, Beijing, China) was used to determine the protein signals.
Cell scratches assay
Following transfection, each group of cells was inoculated in a 6-well plate. The densities and fusion degrees of the waiting cells were 2 × 105/well and > 80%, respectively. The separated cells were aspirated off and reconstituted in basal media (without fetal bovine serum) to culture for 0, 12, 24, and 48 h, respectively. The scratch healing condition was recorded.
EdU assay
EdU tests were used to track cell growth. Following the preparation of NP cells in experimental groups, the cells were inoculated into a 24-well plate at a cell density of 2 × 105. Afterward, 500µL EdU (Sigma, MO, USA) was added to each well, and the cells were incubated at 37 °C for 2 h with 5% CO2. Next, fixed for 15 min with 4% formaldehyde and permeated for 20 min with 0.5% TritonX-100. Following the washing, a 100-liter Apollo reaction mixture was added and incubated in the dark for 30 min. Finally, the results were evaluated under a microscope after 20 min of Hoechst 33,258 staining.
Flow cytometry assay
The apoptosis assay was measured using a flow cytometer and an apoptosis detection kit Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI). The supernatant was transferred to centrifuge tubes for centrifugation, and the NP cells in the logarithmic growth cycle were picked up in centrifuge tubes for centrifugation. The supernatant was removed before the cells were resuspended in the binding buffer. The annexin V-FITC and PI staining solutions were then added, mixed thoroughly, and incubated for 15 min at room temperature in a dark environment. For flow cytometry, the fluorescence intensity of stained cells was measured from the fluorescence intensity, and the apoptotic cells were calculated as a percentage.
Fluorescence in situ hybridization (FISH)
Exiqon QIAGEN developed and manufactured LNA probes with 5’ and 3’-digoxigenin-labeling to confirm miR-106b-5p in NP tissues using FISH (Hilden, Germany). Dilutions and denatured probes were applied dropwise to NP tissue slices from degenerative disc, then hybridized overnight at room temperature. By the following day, it had been taken out, rinsed at room temperature, and dyed with 4, 6-diamino-2-benzene (DAPI). A laser microscope detection was used for detecting fluorescence signals (A1si, Nikon, Tokyo, Japan).
Histological slide evaluation and immunohistochemistry
Tissue was collected from the mouse discs, fixed for one week in 4% formalin, accumulated in 10% EDTA for two weeks, and then paraffin-embedded. The section size was approximately 3 μm and the disc tissue was incised parallel to the median sagittal plane. Sections were histologically examined with Safranin O staining, and scores were assigned using a modified histological grading method based on a recent publication [27]. After hydration of the sections, antigen repair was conducted for 20 min at 100 °C with a 10 mm citric acid buffer, in preparation for immunohistochemistry. Anti-Col II (Abcam: ab34712) and anti-MMP13 (Abcam: ab219620) were used to titrate the sections, which were subsequently incubated overnight at 4 °C. The sections were treated with secondary antibodies for 30 min at room temperature. The color response was subsequently performed using DAB dye, followed by counterstaining with hematoxylin. Finally, images of immunohistochemistry were taken using a microscope (Olympus, Tokyo, Japan).
Statistical analysis
SPSS 23.0 (SPSS, Chicago, USA) was used to conduct all statistical analyses. The mean and standard deviation are used to illustrate numerical data. The statistical differences between the groups were examined using a one-way analysis of variance (ANOVA) with a post hoc test and a student’s t-test, with a value p < 0.05 being classified as statistically significant.
Results
The discovery of miR-106b-5p was up-regulated and positively correlated with the degree of disc degeneration
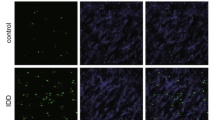
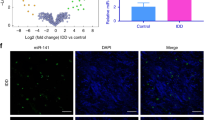
The miRNA microarray analysis of NP tissues from three IDD patients and three controls was used to observe the differential expressions of miRNAs. The results indicated that there were 14 up-regulated and 12 down-regulated miRNAs. These were screened out when the mean change was greater than 5-folds or less than 0.2-fold, with a p-value less than 0.05 (Fig. 1 A). The expression of miR-106b-5p was the most substantially up-regulated among the miRNAs (Supplementary Table 2). The results were validated using qRT-PCR on NP tissue from 212 degenerative samples and 80 control samples. The qRT-PCR research revealed that miR-106b-5p levels in degenerative NP tissues were significantly higher than in controls (Fig. 1B). Similar results were observed in the FISH assay, where miR-106b-5p expression levels were significantly higher in IDD group (Fig. 1 C). Interestingly, there was a positive correlation between miR-106b-5p expression levels and the Pfirrmann IDD classification (n = 45; r = 0.79, p < 0.001; Fig. 1D). Therefore, we theorize that up-regulation of miR-106b-5p may play a crucial role in the development of IDD. The important factor’s screening procedure is depicted in Fig. 1E.

Identification of differently expressed miRNAs in IDD patients’ NP tissues. A 26 differently expressed miRNAs were depicted in a heat map (fold change > 5 or 0.2, p < 0.01). B The qRT-PCR assay was used to examine the different expression levels of miR-106b-5p between the IDD and the control NP samples. C The FISH analysis confirmed the level of miR-106b-5p was higher in NP tissues from IDD patients (The scale bar is 20 μm). D The expression of the miR-106b-5p was found to be positively correlated with the severity grade (Pfirrmann grade) of disc degeneration (n = 45; r = 0.79, p < 0.001). E Microarray-based profiling revealed a miRNAs selection strategy in degenerative NP tissues. *** indicates p < 0.001
The effect of miR-106b-5p overexpression or silence on NP cell
Human NP cells were transfected with miR-106b-5p mimics or inhibitors to create miR-106b-5p overexpression or silencing models for cell function investigations. Fluorograms of green-labeled lentiviruses were used to detect the transfection effect and then validated by qRT-PCR (Fig. 2 A, B). Analysis of the flow cytometric experiments revealed that increased levels of miR-106b-5p promoted apoptosis of NP cells (Fig. 2 C). According to the EdU results, overexpression of miR-106b-5p significantly decreased the proliferation of NP cells (Fig. 2D). In the 48-hour scratch assay, NP cell migration ability was significantly reduced after miR-106b-5p overexpression (Fig. 2E). Next, western blotting was utilized to measure the effects of miR-106b-5p expression on ECM anabolic/catabolic markers. In primary human NP cells transfected with miR-106b-5p mimics, the expression of proteins involved with ECM protection (Col II, Aggrecan) was significantly reduced. In contrast, the opposite phenomenon occurred when transfected with miR-106b-5p inhibitors (Fig. 2 F, G). Overall, we could confirm that overexpression of miR-106b-5p induces apoptosis and inhibits matrix synthesis in NP cells.

Effect of miR-106b-5p on NP cell function. A Transfection of GFP labeled miR-106b-5p into cultured human NP cells. (The scale bar is 50 μm). B The efficacy of miR-106b-5p transfection in human NP cells was shown by qRT-PCR. C Apoptosis rate in human NP cells transfected with miR-106b-5p mimics and inhibitors was measured using flow cytometry. D Cell proliferation was measured in different NP cell groups using the EdU assay. (The scale bar is 50 μm). E The migratory potential of human NP cells transfected with miR-106b-5p mimics/inhibitors was demonstrated in a cell scratch experiment. (The scale bar is 20 μm). F, G Western blotting were used to determine the expression levels of Col II, Aggrecan, MMP13, and ADAMTS5. *** indicates p < 0.001
SIRT2 was a target gene of miR-106b-5p
The miRNA target genes were predicted using five prediction algorithms: miRanda, RNA22, PicTar, TargetScan, and PITA, and the findings were examined using Venn diagrams (Fig. 3A). Cytoscape created the miRNA-mRNA network maps (Fig. 3B). The gene matching score results show that miR-106b-5p was highly conserved across animal species (Fig. 3 C). A luciferase reporter assay confirmed that the luciferase activity of wild-type SIRT2 or mutant SIRT2 was significantly lower than mutant luciferin activity when co-transfected with miR-106b-5p mimics in human NP cells (Fig. 3D). Western blotting showed a significant decrease of SIRT2 protein expression in NP cells transfected with miR-106b-5p mimics (Fig. 3E, F). Our data indicate that miR-106b-5p can suppress the expression of SIRT2 by targeting a specific seed region in the 3’ UTR of SIRT2 mRNA.

Identification of SIRT2 as a target of miR-106b-5p. A, B SIRT2 was predicted as a potential regulatory target for miR-106b-5p. C Sequence alignment revealed that the 3’ UTR of SIRT2 mRNA had high sequence complementarity with miR-106b-5p. D The binding relationship between miR-106-5p and SIRT2 was verified by luciferase reporter assay in human NP cells. E, F The expression of SIRT2 protein was detected by western blotting after transfection of human NP cells with miR-106b-5p mimics or inhibitors. ** indicates p < 0.01 and *** indicates p < 0.001
miR-106b-5p regulated IDD via SIRT2/MAPK/ERK signaling pathway
As shown in Fig. 4 A and B, the differential genome is mostly enriched in the MAPK/ERK signaling pathway according to the Kyoto Encyclopedia of Genes and Genomes (KEGG). Ras, a key factor in the MAPK signaling pathway, can be deacetylated by SIRT2. For this reason, inhibition of SIRT2 can activate the MAPK/ERK signaling pathway. Human NP cells were transfected with miR-106b-5p mimics, inhibitors, or negative controls. The protein expression levels of Ras, ERK1/2, p-ERK1/2, ADAMTS5, and MMP13 were found to be significantly elevated in NP cells with overexpressing miR-106b-5p. Alternatively, the expression of SIRT2, Col II, and Aggrecan proteins was significantly increased in NP cells with silenced miR-106b-5p (Fig. 4 C, D). Furthermore, the effects of SIRT2 overexpression on ADAMTS5, MMP13, Col II, and Aggrecan genes were found to be comparable to those of the miR-106b-5p inhibitor (Fig. 4E, F). Thus, from our findings, miR-106b-5p may induce apoptosis in NP cells and promote IDD progression via SIRT2/MAPK/ERK pathway.

The modulation of miR-106b-5p on the SIRT2/MAPK/ERK signaling pathway. A, B KEGG and GO analyses indicated that the MAPK pathway was abundant in IDD. C, D SIRT2, Ras, ERK1/2, p-ERK1/2, Aggrecan, Col II, MMP13, and ADAMTS5 protein expression levels in miR-106b-5p mimics or inhibitor transfected the human NP cells. E, F Rescue experiments were performed to demonstrate the link between miR-106b-5p and SIRT2 in cultured primary human NP cells. Restoration of SIRT2 expression reversed the inhibition of Aggrecan and Col II protein expression by miR-106b-5p mimics. *** indicates p < 0.001
Down-regulation of miR-106b-5p level prevented IDD development in a mouse model
Following our understanding of the possible pathological mechanism, we sought to further explore the potential therapeutic role of miR-106b-5p in the prevention of IDD. Locally injected the miR-106b-5p mimics (agomiR-106b-5p) or miR-106b-5p inhibitors (antagomiR-106b-5p) in mice models of IDD at 1 day, 1 week and 2 weeks after IDD surgery and compared results to negative controls. The intact discs were obtained after 12 weeks for histological analysis (Fig. 5 A). The Safranin O staining results revealed that the normal basic structure of NP was lost in the disc tissue injected with agomiR-106b-5p, as there were no obvious boundaries between the surrounding fibrous ring and NP. The NP cells were also found to be degenerated or replaced by fibroblast-like phenotype cells. Local administration of antagomir- 106b-5p dramatically alleviated the progression of disc degeneration (Fig. 5B, C). Immunohistochemical studies revealed that antagomiR-106b-5p significantly reduced MMP13 expression but increased Col II expression (Fig. 5D, E). Therefore, this finding suggests that miR-106b-5p inhibitors (antagomiR-106b-5p) could serve as a potential drug for arresting the progression of IDD (Fig. 6).

Down-regulation of miR-106b-5p expression prevents IDD development. A Flowchart of experimental design showed local administration of miR-106b-5p mimics, inhibitors, or negative controls to mice models at 1 day, 1 week, and 2 weeks after IDD surgery. B, C Safranin O staining and histological scoring were used to assess disc degeneration. (The scale bar is 50 μm). D, E Immunohistochemistry of MMP13 and Col II in IDD models treated with miR-106b-5p. (The scale bar is 20 μm). *** indicates p < 0.001

miR-106b-5p inhibitor (antagomiR-106b-5p) is a potential therapeutic target to attenuate the progression of IDD. This schematic diagram shows that miR-106b-5p induces IDD through the stimulation of NP cell catabolism and apoptosis via SIRT2/ MAPK/ERK signaling pathway. Furthermore, the miR-106b-5p inhibitor (antagomiR-106b-5p) alleviates the degenerative process of intervertebral disc in IDD mouse model
Discussion
It has been well documented that miRNAs play critical roles in IDD development. To date, no miRNA has entered into clinical trial for rescuing IDD [8, 28]. To the best of our knowledge, this is the first preclinical study to identify the therapeutic significance of miR-106b-5p in the treatment of IDD. Based on miRNA microarray expression profiles, miR-106b-5p expression was found to be up-regulated in degenerated NP tissues compared to control NP tissues. Furthermore, it was observed that the severity of IDD was also positively correlated with the differential expression of miR-106b-5p. Thus miR-106b-5p was selected as a target miRNA for further investigation. In vitro cell function tests revealed that miR-106b-5p overexpression contributed to an imbalance in ECM anabolic and catabolic metabolism. Fundamentally, miR-106b-5p could directly target SIRT2 mRNA, while SIRT2 overexpression could rescue the degradation of ECM caused by miR-106b-5p. Through these studies, it was revealed that miR-106b-5p and SIRT2 might be indispensable factors in the progression of IDD.
SIRT2 is a nicotinamide adenine dinucleotide-dependent (NAD+) histone deacetylase that belongs to the sirtuin family of proteins [29, 30]. Current research has confirmed that SIRT2 plays an important role in abnormal physiological activities such as senescence, apoptosis, and inflammation. These abnormal physiological activities are thought to be critical in the development of IDD [31, 32]. The role of MAPK as an intracellular signal transduction pathway has been proven to play a critical regulatory role in the development of IDD [33]. In addition, new evidence shows that SIRT2 can regulate the MAPK signaling pathway directly through deacetylation [34]. Nevertheless, the exact molecular mechanism through which SIRT2 deacetylase regulates IDD in the MAPK/ERK signaling pathway is unknown. This study identified that miR-106b-5p can regulate NP cell function via direct binding to the SIRT2-3’ UTR. Ras is an important upstream regulator of the MAPK/ERK signaling pathway that can be regulated by the deacetylation of SIRT2 [35,36,37]. Therefore, it is hypothesized that SIRT2 can regulate the MAPK/ERK signaling pathway through deacetylation, which is involved in the progression of IDD. In this study, when miR-106b-5p was overexpressed in NP cells, the expression and acetylation of SIRT2 were reduced, which ultimately contributed to a significant increase in Ras and MAPK/ERK signaling protein expression. Conversely, overexpression of SIRT2 counteracted the effects of miR-106b-5p overexpression on Col II, Aggrecan, ADAMTS5, and MMP13 proteins. These studies further elucidated the role of miR-106b-5p in MAPK signaling and represent the current research avenues for the investigation of miR-106b-5p in the regulation of IDD. Nevertheless, even though miR-106b-5p regulates SIRT2, further in-depth mechanisms should be explored in future studies.
miRNA-based therapy is a very attractive treatment strategy of IDD [38]. Recent preclinical studies have found that miRNA-based therapeutic approach is feasible and effective through direct injection of miRNA vectors into the intervertebral disc [8, 9]. In this study, we conducted in vivo experimentation with the AF acupuncture-induced IDD mouse model to investigate the potential therapeutic significance of miR-106b-5p. As reported, the local injection of agomiR-106b-5p resulted in increased NP cell apoptosis, higher histological scores, and augmented production of catabolic markers (MMP13). IDD progression was alleviated by silencing miR-106b-5p after local injection of antagomiR-106b-5p in the IDD model. These data indicate that miR-106b-5p inhibitors can arrest the development of IDD by remodeling the balance between ECM anabolic/catabolic factors. Our study might provide a new theoretical foundation for the clinical translation of miR-106b-5p for IDD treatment.
The therapeutic potential of miRNAs has been demonstrated in multiple preclinical studies of IDD [39]. Due to the role of miRNA in many disc degenerative process, such as cell proliferation, apoptosis, homeostasis and metabolism, it is noteworthy that miRNA-based therapeutics is a good candidate for IDD [8, 9, 40, 41]. However, there are still many practical challenges in miRNA-based therapeutics, including safety, efficacy, target selection, and delivery technologies. For instance, local delivery to the intervertebral disc tissue is a major obstacle that will always complicate the path to clinical application [28]. Optimal miRNA delivery in vivo is always accessible to the target cell without its degradation by internal nucleases. However, the number of pre-clinical or clinical trials in this regard is very low. Moreover, the mechanism of action of miRNA in targeting different signaling pathways is still poorly understood. Clarifying and overcoming these challenges are extremely important for laying a solid foundation for the clinical translation of miRNA-based therapy.
There are several limitations in our study. First, the sample size of this study may be relatively small. It is vital to verify the phenotypic specificity of miR-106b-5p in the large-scale studies. Second, potential off-target effect of antagomiR-106b-5p in vivo remains undetermined. Third, the toxicity of intradiscal delivery of miR-106b-5p occurring in surrounding tissues is warranted in future investigation.
In summary, miRNA microarray analyses showed that miR-106b-5p expression was significantly up-regulated in degenerative human NP samples. This result was verified by qRT-PCR and FISH. Gain- and loss-of-function experiments revealed that miR-106b-5p promoted IDD by inducing NP cell apoptosis and ECM degradation via the SRIT2/MAPK/ERK pathway. Furthermore, intradiscal delivery of antagomiR-106b-5p significantly attenuated IDD development in the animal model. Our findings indicate that targeting miR-106b-5p in interverbal disc has therapeutic effects on IDD.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Luoma K, Riihimaki H, Luukkonen R, Raininko R, Viikari-Juntura E, Lamminen A (2000) Low back pain in relation to lumbar disc degeneration. Spine (Phila Pa 1976) 25:487–492. https://doi.org/10.1097/00007632-200002150-00016
Andersson GB (1999) Epidemiological features of chronic low-back pain. Lancet 354:581–585. https://doi.org/10.1016/S0140-6736(99)01312-4
Samartzis D, Karppinen J, Mok F, Fong DY, Luk KD, Cheung KM (2011) A population-based study of juvenile disc degeneration and its association with overweight and obesity, low back pain, and diminished functional status. J Bone Joint Surg Am 93:662–670. https://doi.org/10.2106/JBJS.I.01568
Airaksinen O, Brox JI, Cedraschi C et al (2006) Chap. 4. European guidelines for the management of chronic nonspecific low back pain. Eur Spine J 15 Suppl 2S192–300. https://doi.org/10.1007/s00586-006-1072-1
Global Burden of Disease Study C (2015) Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386:743–800. https://doi.org/10.1016/S0140-6736(15)60692-4
Radcliff KE, Kepler CK, Jakoi A et al (2013) Adjacent segment disease in the lumbar spine following different treatment interventions. Spine J 13:1339–1349. https://doi.org/10.1016/j.spinee.2013.03.020
Zhou X, Chen L, Grad S et al (2017) The roles and perspectives of microRNAs as biomarkers for intervertebral disc degeneration. J Tissue Eng Regen Med 11:3481–3487. https://doi.org/10.1002/term.2261
Ji ML, Jiang H, Zhang XJ et al (2018) Preclinical development of a microRNA-based therapy for intervertebral disc degeneration. Nat Commun 9:5051. https://doi.org/10.1038/s41467-018-07360-1
Jiang H, Moro A, Wang J, Meng D, Zhan X, Wei Q (2021) MicroRNA-338-3p as a novel therapeutic target for intervertebral disc degeneration. Exp Mol Med 53:1356–1365. https://doi.org/10.1038/s12276-021-00662-3
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. https://doi.org/10.1016/j.cell.2009.01.002
He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531. https://doi.org/10.1038/nrg1379
Ali SA, Peffers MJ, Ormseth MJ, Jurisica I, Kapoor M (2021) The non-coding RNA interactome in joint health and disease. Nat Rev Rheumatol 17:692–705. https://doi.org/10.1038/s41584-021-00687-y
Miyaki S, Asahara H (2012) Macro view of microRNA function in osteoarthritis. Nat Rev Rheumatol 8:543–552. https://doi.org/10.1038/nrrheum.2012.128
Jing W, Jiang W (2015) MicroRNA-93 regulates collagen loss by targeting MMP3 in human nucleus pulposus cells. Cell Prolif 48:284–292. https://doi.org/10.11 11/cpr.12176
Wang HQ, Yu XD, Liu ZH et al (2011) Deregulated miR-155 promotes Fas-mediated apoptosis in human intervertebral disc degeneration by targeting FADD and caspase-3. J Pathol 225:232–242. https://doi.org/10.1002/path.2931
Cazzanelli P, Wuertz-Kozak K (2020) MicroRNAs in Intervertebral Disc Degeneration, Apoptosis, Inflammation, and Mechanobiology. Int J Mol Sci 21. https://doi.org/10.3390/ijms21103601
Henry N, Clouet J, Le Bideau J, Le Visage C, Guicheux J (2018) Innovative strategies for intervertebral disc regenerative medicine: From cell therapies to multiscale delivery systems. Biotechnol Adv 36:281–294. https://doi.org/10.1016/j.biotechadv.2017.11.009
Tan F, Wang D, Yuan Z (2020) The Fibroblast-Like Synoviocyte Derived Exosomal Long Non-coding RNA H19 Alleviates Osteoarthritis Progression Through the miR-106b-5p/TIMP2 Axis. Inflammation 43:1498–1509. https://doi.org/10.1007/s10753-020-01227-8
Yang Y, Fang S (2017) Small non-coding RNAs-based bone regulation and targeting therapeutic strategies. Mol Cell Endocrinol 456:16–35. https://doi.org/10.101 6/j.mce.2016.11.018
Griffith JF, Wang YX, Antonio GE et al (2007) Modified Pfirrmann grading system for lumbar intervertebral disc degeneration. Spine (Phila Pa 1976) 32:E708–712. https://doi.org/10.1097/BRS.0b013e31815a59a0
Xia X, Guo J, Lu F, Jiang J (2015) SIRT1 Plays a Protective Role in Intervertebral Disc Degeneration in a Puncture-induced Rodent Model. Spine (Phila Pa 1976) 40:E515–524. https://doi.org/10.1097/BRS.0000000000000817
Ji ML, Zhang XJ, Shi PL et al (2016) Downregulation of microRNA-193a-3p is involved in invertebral disc degeneration by targeting MMP14. J Mol Med (Berl) 94:457–468. https://doi.org/10.1007/s00109-015-1371-2
Showalter BL, Beckstein JC, Martin JT et al (2012) Comparison of animal discs used in disc research to human lumbar disc: torsion mechanics and collagen content. Spine (Phila Pa 1976) 37:E900–907. https://doi.org/10.1097/BRS.0b013e31824d911c
Beckstein JC, Sen S, Schaer TP, Vresilovic EJ, Elliott DM (2008) Comparison of animal discs used in disc research to human lumbar disc: axial compression mechanics and glycosaminoglycan content. Spine (Phila Pa 1976) 33:E166–173. https://doi.org/10.1097/BRS.0b013e318166e001
Wang F, Wu XT, Zhuang SY et al (2011) Ex vivo observation of human nucleus pulposus chondrocytes isolated from degenerated intervertebral discs. Asian Spine J 5:73–81. https://doi.org/10.4184/asj.2011.5.2.73
Bu D, Luo H, Huo P et al (2021) KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res 49:W317–W325. https://doi.org/10.1093/nar/gkab447
Masuda K, Aota Y, Muehleman C et al (2005) A novel rabbit model of mild, reproducible disc degeneration by an anulus needle puncture: correlation between the degree of disc injury and radiological and histological appearances of disc degeneration. Spine (Phila Pa 1976) 30:5–14. https://doi.org/10.1097/01.brs.0000148152.04401.20
Roh EJ, Darai A, Kyung JW et al (2021) Genetic Therapy for Intervertebral Disc Degeneration. Int J Mol Sci 22. https://doi.org/10.3390/ijms22041579
Vaquero A, Scher MB, Lee DH et al (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev 20:1256–1261. https://doi.org/10.1101/gad.1412706
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13. https://doi.org/10.1042/BJ20070140
Zhang GZ, Deng YJ, Xie QQ et al (2020) Sirtuins and intervertebral disc degeneration: Roles in inflammation, oxidative stress, and mitochondrial function. Clin Chim Acta 508:33–42. https://doi.org/10.1016/j.cca.2020.04.016
Zhao L, Cao J, Hu K et al (2020) Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis 11:927–945. https://doi.org/10.14336/AD.2019.0820
Zhang HJ, Liao HY, Bai DY, Wang ZQ, Xie XW (2021) MAPK /ERK signaling pathway: A potential target for the treatment of intervertebral disc degeneration. Biomed Pharmacother 143:112170. https://doi.org/10.1016/j.biopha.2021.112170
She DT, Wong LJ, Baik SH, Arumugam TV (2018) SIRT2 Inhibition Confers Neuroprotection by Downregulation of FOXO3a and MAPK Signaling Pathways in Ischemic Stroke. Mol Neurobiol 55:9188–9203. https://doi.org/10.1007/s12035-018-1058-0
Jing H, Zhang X, Wisner SA et al (2017) SIRT2 and lysine fatty acylation regulate the transforming activity of K-Ras4a. Elife 6 https://doi.org/10.7554/eLife.32436
Li Y, Zhang M, Dorfman RG et al (2018) SIRT2 Promotes the Migration and Invasion of Gastric Cancer through RAS/ERK/JNK/MMP-9 Pathway by Increasing PEPCK1-Related Metabolism. Neoplasia 20:745–756. https://doi.org/10.1016/j.neo.2018.03.008
McCubrey JA, Steelman LS, Chappell WH et al (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773:1263–1284. https://doi.org/10.1016/j.bbamcr.2006.10.001
Li Z, Rana TM (2014) Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov 13:622–638. https://doi.org/10.1038/nrd4359
Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16:203–222. https://doi.org/10.1038/nrd.2016.246
Le Moal B, Lepeltier E, Rouleau D et al (2022) Lipid nanocapsules for intracellular delivery of microRNA: A first step towards intervertebral disc degeneration therapy. Int J Pharm 624:121941. https://doi.org/10.1016/j.ijpharm2022.121941
Wu T, Jia X, Zhu Z et al (2022) Inhibition of miR-130b-3p restores autophagy and attenuates intervertebral disc degeneration through mediating ATG14 and PRKAA1. Apoptosis 27:409–425. https://doi.org/10.1007/s10495-022-01725-0
Acknowledgements
The authors thank all the participants in this study.
Funding
This work was supported by the National Natural Science Foundation of China (81860406), Guangxi Natural Science Foundation (2018GXNSFAA281127), and The Medical Excellence Award Funded by the Creative Research Development Grant from The First Affiliated Hospital of Guangxi Medical University.
Author information
Authors and Affiliations
Contributions
HJ and Q-JW conceived and designed the experiments; HJ and AM contributed to critical revision of the manuscript; D-HM, W-YC and CP performed the experiments; Y-WG, J-QW and K-XY analyzed and interpreted the data and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
The histological study of surgical samples was approved by the ethics committee of The First Affiliated Hospital of Guangxi Medical University (2018-KY-NSFC-025).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dihua Meng and Weiyou Chen are equal contribution to this work
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meng, D., Chen, W., Pan, C. et al. Exploration of microRNA-106b-5p as a therapeutic target in intervertebral disc degeneration: a preclinical study. Apoptosis 28, 199–209 (2023). https://doi.org/10.1007/s10495-022-01773-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01773-6