
Abstract
Resveratrol, a naturally occurring polyphenolic antioxidant, is a potential chemoprophylactic agent for various cancers, including colorectal cancer. Although emerging evidence continually suggests that a number of resveratrol derivatives may be better cancer chemopreventive candidates than resveratrol, studies on the mechanism of action of these derivatives are limited. This is the first study which investigates the mechanism underlying the cytotoxic effect of a synthesized resveratrol analogue, (E)-N-(2-(4-methoxystyryl) phenyl) furan-2-carboxamide (CS) on colorectal cancer. Previously, our group reported a series of synthesized resveratrol analogues, which showed cytotoxicity against a panel of cancer cell lines, in particular on colon cancer cells. In this study, we further discovered that CS also exerts a potent suppressive effect on HCT116 colorectal cancer cells. In contrast, normal colon cells (CCD-112 Con) were not sensitive to CS up to 72 h post treatment. CS caused cytotoxicity in HCT116 cells through several apoptotic events including activation of the Fas death receptor, FADD, caspase 8, caspase 3, caspase 9, and cleaved PARP, which occurred alongside cell cycle arrest from the up-regulation of p53 and p21. The results show that CS causes apoptosis via the activation of an extrinsic pathway leading to caspase activation and cell cycle arrest from activated p53. These findings suggest that CS may be a potential candidate for development as an anti-tumor agent in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is the second leading cause of death worldwide. Colorectal cancer in particular is one of the most malignant neoplasia, being the third most common type of cancer among men and second among women [38, 40]. Improvements in surgery, adjuvant therapy, dietary plans, and screening programs have facilitated an overall decline in the mortality rate of colon cancer. However, the inherent and acquired drug resistance limits many chemotherapeutic agents from treating colorectal cancer, as they are often associated with severe, dose limiting, and systemic toxicities [15, 34].
Apoptosis is a continuous physiological mechanism for non-inflammatory, programmed cell death and is frequently investigated as the mechanism of action for anti-tumor agents [18]. Two statutory pathways have been shown to regulate caspase-dependent apoptosis—the extrinsic “death-receptor-mediated” and—the intrinsic “mitochondrial-mediated” pathways [13]. The extrinsic pathway is initiated by the binding of a death receptor ligand, which activates the tumor necrosis factor (TNF) superfamily of death receptors and downstream caspase-8. For the intrinsic pathway, assaults on the mitochondria trigger the release of pro-apoptotic factors such as cytochrome-c and downstream caspase-9. These two pathways converge in the activation of effector caspases such as caspases-3 and -7 [27, 28]. The cell morphological changes of an apoptotic cell include externalization of phosphatidylserine (PS) of the cell membrane, membrane blebbing, chromatin condensation, and nuclear DNA fragmentation [37].
The cell cycle has safeguard mechanisms that control the rate of cell replication. Homeostasis between cell death and replication must be balanced to maintain various types of cells in tissues. This mechanism has been a major target for the development of new chemotherapeutic agents, as defects in cell cycle checkpoints will lead to uncontrolled cell proliferation [7, 9]. There is evidence showing the relationship between apoptosis and cell cycle through some regulatory proteins in the cells where the manipulation of these regulatory proteins may prevent or induce apoptosis [31]. Of note, the p53 protein has been identified to be pivotal in deciding cellular growth arrest or apoptosis [32]. The p53 protein is particularly able to regulate two gene targets those, which encode proteins that act through the receptor-mediated signaling, and those that regulate apoptotic effector proteins [30]. A chemotherapeutic agent can activate various apoptotic and cell cycle arrest pathways and thus cause death to cancer cells. Therefore, new compounds that target colorectal cancer should be investigated to meet the needs of novel improved therapeutic agents for colorectal cancer.
Interest in the pharmacological effects of bioactive compounds for cancer treatment and prevention has increased significantly over the past two decades. Natural agents derived from plants and other natural resources can be useful as a complementary therapy for cancer patients. Furthermore, there is a need to develop more chemotherapeutic options from natural resources with reduced side effects to improve current chemotherapy [39]. Amongst many bioactive compounds, resveratrol (3,5,4′-trihydroxystilbene), a naturally occurring non-flavonoid polyphenol obtained from several plant resources such as grapes, mulberries, and peanuts, has been shown to exhibit a range of health benefits such as anti-oxidant, anti-inflammatory, cardio-protective, and anti-tumor potential properties [2, 33]. Unfortunately, high doses of resveratrol are required to induce apoptosis in cancer cells. In addition, resveratrol’s biological activity is limited by its photosensitivity and metabolic instability [5]. Thus, recent studies on resveratrol have shifted to synthetic analogues with improved anti-tumor activity. Our group previously designed, synthesized, and investigated the cytotoxicity of a series of resveratrol analogues. The compound 6d or CS was shown to be most potent on colon HT-29 cells with an IC50 of 11.40 µM [25]. Therefore, in this report, we seek to understand how CS exerts its apoptotic and cell cycle arrest on the human colon cancer cell line, HCT116.
Materials and methods
(E)-N-(2-(4-methoxystyryl) phenyl) furan-2-carboxamide
(E)-N-(2-(4-methoxystyryl) phenyl) furan-2-carboxamide (CS) was synthesized and purified according to a previously published method. CS is obtained as a white powder. The 1H and 13C nuclear magnetic resonance (NMR) (JEOL JNM-LA 400 and JEOL ECA-400) spectra of the compound obtained in the CDCl3 solution were in agreement with a previous report [25]. The 1H NMR (CDCl3, 400 MHz) δ: 7.14 (br s, 1H), 6.94 (td, J = 1.4, 8.2 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 7.16–7.05 (m, 3H), 6.81 (d, J = 16.5 Hz, 1H), 7.04–7.10 (m, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 2.10 (s, 3H, CH3); 13C NMR (CDCl3, 100 MHz) 139.6, 112.9, 162.0, 113.1, 130.2, 122.8, 130.3, 125.3, 130.4, 134.6, 124.7, 128.5, 125.8, 126.7, 24.2 (CH3), 168.6; HRMS (+ ESI) [M+H]+: 256.1810, and C16H15FNO require 256.1138. Figure 1 shows the structure of CS.

Chemical structure of (E)-N-(2-(4-methoxystyryl) phenyl) furan-2-carboxamide (CS)
Cell culture
HT-29 cells, HCT116, and CCD-112 Con were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). HT-29 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM). The HCT116 cells in McCoy’s 5a Modified Medium and CCD-112 Con in Eagle’s Minimum Essential Medium (EMEM), were all supplemented with 10% fetal bovine serum. The cells were sub-cultured into culture flasks and incubated at 37 °C in a humidified atmosphere containing 5% CO2 until approximately 70–80% confluence. The cells were passaged every 3–4 days. All cell lines were routinely tested for contamination and mycoplasma infection [20].
Cytotoxicity assay
To determine the number of viable cells upon treatment of CS, CellTiter 96®AQueous One (Promega, Madison, WI, USA) cytotoxicity assay was used. Briefly, the cells were seeded at 1 × 104 for HCT116, 5 × 103 for HT29, 2 × 104 cells per well for CCD-112 Con, and allowed to grow overnight in clear 96-well microplates. The cells were treated with the compound or standard drug standards at a concentration of 0.4–200 µM dissolved in dimethyl sulfoxide, (DMSO) for 24, 48, and 72 h. For the negative control, the cells were treated with a DMSO (0.1% v/v) solvent only. After the incubation period, the cells were refreshed with 100 µL of DMEM media followed by the addition of 20 µL per well of [3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)]. Then, the plates were incubated at 37 °C for 1–2 h in a humidified, 5% CO2 atmosphere. Next, the absorbance was obtained using a microplate reader set at 490 nm with 690 nm as the background wavelength (Infinite® 200 Pro, Tecan, Mӓnnedorf, Switzerland). The inhibition concentration at 50% (IC50) and statistical significance were determined using dose-response curves and the Student’s T test (p < 0.05) via the Prism 5.02 software (GraphPad Software Inc., La Jolla, CA, USA) [20].
Caspase activity
The activities of caspase-3/7, -8 and -9 were measured using Caspase-Glo® 3/7,8, and 9 assay kits (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, the cells were plated over-night in a white 96-well microplate. A total of 1 × 104 cells were seeded per well and treated with CS (6 µM) for 3, 7, 18, 24, and 30 h, with or without inhibitors (caspase-8: Z-IETD-FMK; caspase-9: Z-LEHD-FMK; caspase-3: Z-DEVD-FMK). After treatment, 100 µL of each caspase substrate was added and incubated at room temperature for 30 min. Upon activation of caspase in the apoptotic cells, the substrate will give a luminescence signal after cleavage of the aminoluciferin-labelled synthetic tetrapeptide. The luminescence is then detected using a Tecan Infinite® 200 Pro (Tecan, Männedorf, Switzerland) microplate reader [20].
Apoptosis assay
Annexin V is a phospholipid-binding protein and has a high affinity towards PS, a property that is used to determine the degree of apoptosis induced by CS on HCT116 cells. The cells were seeded at 1 × 105 cells per ml in a 25 cm2 flask overnight before treatment with CS for 24 h at various concentrations. After treatment, the cells were incubated with FITC-annexin V and propidium iodide (PI) (BD Biosciences) in a binding buffer for 15 min in the dark and in ice-cold conditions. The stained cells were measured using a FACS Canto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed with BD FACS Diva version 8.0.1 (Verity Software House Inc., Topsham, ME, USA) [20].
Cell cycle analysis
To determine the cell cycle status following treatment with CS, staining of the DNA was performed and analyzed via the flow cytometry technique. In brief, HCT116 cells (2 × 105) were cultured in a 25 cm2 flask and incubated for 24 h, after which the cells were treated with either CS (6 µM) or DMSO (0.1%, v/v) for 24 and 48 h. After treatment, the cells were collected and fixed with cold 70% ethanol. Prior to analysis, the cells were washed twice with phosphate buffer saline (PBS) and incubated with RNase A and followed by propidium iodide for 10 min each. The cells were analyzed on a FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA). The cell cycle phase distribution and proportion of apoptotic cells (percentage of sub-G0 phase) were analyzed from the DNA histogram using ModFit version 3.3 (Verity Software House Inc., Topsham, ME, USA) [20].
Transferase-UTP nick-end labeling (TUNEL) assay
Cleaved DNA strands can be identified by labeling the free 3′-OH terminal of the DNA with terminal deoxynucleotidyltransferase (TdT), which was detected using a Nikon Eclipse TE2000-S Microscope with MetaMorph software 7.6.0.0 (Nikon, TOKYO, JAPAN). HCT116 cells were plated on glass slides and treated with CS for 6, 12, 18, and 24 h. The pre-treated cells were fixed, permeabilized, labeled, and stained according to the manufacturer’s protocol. The slides were mounted using Vectashield® with DAPI and observed. Localized green fluorescence of apoptotic cells and DAPI-stained nuclei were taken from the same field at ×100 magnification [20].
Apoptosis array analysis
In order to study the signal transduction pathway of CS treated HCT116 cells, an apoptosis array was performed using a Ray-Bio Human Apoptosis Antibody Array G Series (RayBiotech, Norcross, GA, USA). HCT116 cells were plated in 24 well plates and treated with either CS (6 µM) or DMSO (0.1%, v/v) for 6, 12, 18, and 24 h. Cell lysate was collected using a cell lysis buffer the kit. The glass was incubated in a blocking buffer for 30 min. After removing the blocking buffer, the cell lysate was added and incubated overnight at 4 °C. After decanting the sample, the antibody array was washed and stained according to manufacturer protocol. The antibody array chip was scanned using a LI-COR imaging system (Lincoln, Nebraska, USA). The intensity of the array chip was quantified and analyzed with ImageStudio™ Lite software (LI-COR Biosciences, Lincoln, Nebraska, USA) [20].
Western blot analysis
Whole cell lysates were prepared from CS treated cells; cell monolayers were rinsed with cold PBS and then lysed with a cell lysis buffer with protease, phosphatase inhibitors, and EDTA cocktails (Pierce Thermo Fisher Scientific, Waltham, MA, USA). The cell suspensions were centrifuged at 14,000×g for 10 min at 4 °C and the protein concentration was determined using a BCA protein assay kit (Pierce Thermo Fisher Scientific, Waltham, MA, USA). An equal volume of total protein was mixed with the sample loading buffer and separated by 10–12% SDS-polyacrylamide gel and then transferred to a PVDF membrane for the western blot analysis. Immunoblotting was performed using primary antibodies (1:1000) against β-actin, Fas, caspase-8, caspase-3, caspase-9, cleaved PARP, FADD, p53, p21Waf1/Cip1, Bax, and Bcl-xL (Cell Signaling Technologies, Danvers, MA, USA). The membranes were incubated at 4 °C overnight on an orbital shaker and then washed with tris-buffered saline with Tween 20 (TBST) three times for 10 min each. After that, the membranes were incubated with goat anti-rabbit secondary antibody (1:1000) for 4 h at room temperature and developed using the WesternBright ECL kit (Advansta, CA, USA). Finally, the membranes were analyzed and quantified using a UVP Gel documentation system (UVP, CA, USA) [20].
ROS and superoxide assay
For the determination of intracellular ROS levels, the cells were grown in 96-well microplate and treated with CS (6 µM) or DMSO (0.1%, v/v) for 30 and 60 min. After incubation, the cells were stained with a ROS/Superoxide detection kit (Abcam, San Francisco, USA) for 60 min at 37 °C in a CO2 incubator. The fluorescence intensity of the cell was analyzed using a FACS Canto II flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA) [20].
Statistical analysis
All data represent at least three independent experiments and expressed as means ± standard deviation (SD). For statistical analysis, a one-way analysis of variance (ANOVA) was used for the comparison of the differences among more than two groups via the GraphPad Prism software (version 5.0; GraphPad Software Inc., San Diego, CA, USA) [20].
Results
Inhibitory effects of CS on colon cancer cells
To evaluate the cytotoxicity of CS on HCT116, HT29, and CCD-112Con cells, a CellTiter 96®AQueous One solution cytotoxicity assay was performed. The results demonstrated no cytotoxicity on normal colon cells, CCD-112 Con (Table 1). However, CS was cytotoxic towards cancer cells in a time- and dose-dependent manner on HCT116 and HT29 cells. Therefore, our data suggests that CS is selective towards colon cancer cells, HCT116 and HT29. Since the IC50 values are the lowest in HCT116 cell lines, further studies were conducted using this cell line.
Apoptosis by CS in HCT116 colon cancer cells
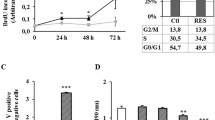
To determine whether or not CS induces cell death via apoptosis or necrosis in HCT116 colon cancer cells, annexin-V-FITC and propidium iodide (PI) dual labeling was performed on CS treated cells at 3, 6, and 12 µM for 24 h. The results show a gradual increase in early apoptotic cells (annexin V positive only) from 15.77 ± 4.34 to 16.27 ± 1.00 and 17.43 ± 3.23%, respectively (Fig. 2).This trend is also observed in late apoptotic cells (annexin V and PI positive), where both increased from 21.93 ± 1.66 to 33.07 ± 4.15% and 37.30 ± 4.25% compared to the negative control (cells treated with 0.1% v/v DMSO). The percentage of necrotic cells (annexin V negative and PI positive) was minimal (~ 4%) throughout the treatment concentrations. Hence, the results show that CS inhibited HCT116 cell growth by inducing apoptosis.

Annexin V/PI double staining assay. a Dot plots of untreated and CS treated HCT116 cells (3–12 µM). b The population of cells (total of 100%) that are viable, in the early and late apoptosis, and necrosis state. The percentage is represented as the mean ± SD of three independent experiments and the symbol * denotes significant difference (p < 0.05) compared to the untreated control
CS halts G2/M cell cycle arrest
Cell cycle regulation is an important factor in the development of cancer. Furthermore, disruption of the cell cycle may lead to the formation of cancer cells. Thus, the cell cycle machinery is the main objective in limiting cancer progression and development [35]. Therefore, to determine the effect of CS on cell cycle, treated cells were labeled with PI in order to measure the cell DNA content in relation to the phases of the cell cycle using a flow cytometry analysis. CS treated cells were significantly reduced in the G0/G1 phase, with accumulation of cells in the G2/M phase after 24 and 48 h post-treatment (close to 80% of cells in G2/M), suggesting that CS causes cellular arrest in the G2/M phase [7] (Fig. 3).

Effects of CS on HCT116 cell cycle progression. a Flow cytometer histograms of untreated and treated (6 µM) cells. b Percentage of cells in different phases of cell cycles after treatment with CS for 24 and 48 h, significant cell cycle arrest at G2/M phase was observed. All data are expressed as the mean ± SD of triplicate measurements. The symbol * denotes significant increase (p < 0.05) compared to the untreated control
Reactive oxygen species (ROS) generation by CS
The production of reactive oxygen species (ROS) is known to activate a series of adaptive cellular responses ranging from growth arrest to apoptosis [22]. In this experiment, the ROS assay monitors the ROS production in live cells upon treatment of CS after 30 and 60 min using flow cytometry analysis. Flow cytometry detects the number of cells stained with oxidative stress dye (green channel, x-axis) and superoxide dye (orange channel, y-axis). The cells, which produce ROS species, exhibit positive staining on both oxidative and superoxide dyes. The results were analyzed according to the percentage of cells in each quadrant of the dot plot. As shown in Fig. 4, treatment with 6 µM CS significantly increased the level of ROS generated by HCT116 cells over time. Additionally, we monitor the ROS production in HT-29 and CCD-112 Con treated cells as well. We observe the similar ROS generation in CS treated HT-29 cells but minimal ROS generation in CCD-112 Con normal colon cells (below 5%).


ROS release in HCT-116, HT-29 and CCD-112 Con cells. a Flow cytometer dot plots of oxidative stress and superoxide detection. b The percentage increase in oxidative stress and superoxide generation in cell after exposure to CS for 30 and 60 min. All the data are expressed as the mean ± SD of triplicate measurements and the symbol * denotes significant increase (p < 0.05) compared to the untreated control
CS treated in HCT116 cells causes caspase activation
Caspase is activated after site-specific cleavage of the pro-forms that will degrade cellular proteins in apoptotic cells [36]. In order to study whether or not caspases are involved in CS induced apoptosis, caspase-3/7, -8, and -9 activities were quantified in HCT116 treated cells. In this study, caspase -3/7, -8, and -9 were significantly elevated compared to the control after 18, 24, and 30 h of incubation (Fig. 5). High levels of caspase-8 (14-fold over control) were detected at 18 h post-treatment. Caspase-9 also exhibited a two-fold increase in elevation over the control. This suggests that CS activates caspase-8 after 18 h of treatment. In addition, at 24 h, caspase-9 was also activated, which leads to the accumulation of caspase-3/7 at a later point in time. To further confirm the activation of caspases by CS, inhibitors of each specific caspases, caspase-3 inhibitor (Z-DEVD-FMK), caspase-8 inhibitor (Z-IETD-FMK), and caspase-9 inhibitor (Z-LEHD-FMK), were added to the CS treated cells, and the subsequent activation of each respective caspase was observed. In the presence of inhibitors, activation of caspase-3, -8, and -9 by CS was ablated in HCT116 cells.

Effect of CS on caspase activation. Fold increase of a caspase-3/7, b caspase-8, and c caspase-9 in HCT116 cells treated with 6 µM of CS as compared to the untreated control. The caspase activities were determined in the presence or absence of specific caspase inhibitors at various time points. All results are obtained from three independent experiments. The symbol * indicates significant increase (p < 0.05) compared to 0 h or non inhibitor treated
The apoptosis pathway of CS
In order to identify the apoptosis pathway(s) induced by CS on HCT116 cells, a Ray-Bio Human apoptosis antibody array was performed. The cells were treated for 6, 12, 18, and 24 h alongside DMSO (0.1%, v/v) for each time point as the control. The results reveal significant increase in protein expression of activated Fas, p21, and p53 in cells treated with CS at 18 h onwards compared to the untreated control (Fig. 6).

Expression of apoptotic protein signals from the apoptosis array study in CS treated HCT116 cells at 6, 12, 18, and 24 h. a Expression of apoptotic proteins detected via human apoptosis antibody array. b Fold changes of apoptosis signals in comparison to the untreated control. The symbol * denotes a significant increase (p < 0.05) compared to the untreated control
Effects of CS on the death domain, caspases, and tumor suppressor protein expressions
In order to validate the apoptotic pathways obtained from the human apoptosis antibody array and caspase assay, we analyzed the expression profile of the death domain, caspase, and tumor suppressor proteins using the western blot approach. The whole cell lysate of CS treated cells was collected after 6, 12, 18, and 24 h incubation. As shown in Fig. 7, CS increased the cleaved caspases -3, -8, and -9 (the activated form of caspases) in a time-dependent fashion. Notably, these observations are consistent with the results obtained in the luminescence caspase assay. Additionally, Fas and FADD protein expressions were evidenced over time. This further suggests that the compound activates the death domains via the Fas activation pathway, which is in agreement with the apoptosis array results. The p53 and p21waf1/cip1 expressions were also increased upon treatment of CS in HCT116 cells. The up-regulation of the Bax and expression of Bcl-xL was also consistent, as per the p53 and p21waf1/cip1 findings [11, 12]. Cleaved PARP was distinct, confirming the occurrence of late apoptosis in HCT116 cells attributed to CS treatment.


Western blot analyses of apoptosis-associated protein after CS treatment. HCT116 cells were treated with CS for 6, 12, 18, and 24 h and probed for the expression. a Fas and FADD. b Caspase-3, -8, -9 (and cleaved). c p21, p53, PARP (and cleaved), Bax and Bcl-xL. β-actin was used as the housekeeping protein. Quantification of the proteins was performed via densitometric analysis of the western blot technique and normalized to the internal loading control. Data consists of the mean ± SD from at least three independent experiments; values are relative to untreated control cells. (***p ≤ 0.001 compared with control cells)
DNA fragmentation in HCT116 due to apoptosis induced by CS
A transferase-UTP nick-end labeling (TUNEL) assay was performed to evaluate CS-induced apoptosis in HCT116 cells. The hallmarks of apoptotic cells are cell shrinkage with nuclear chromatin condensation and DNA strand breakage [8]. As shown in Fig. 8, the untreated cells showed negative staining, whereas treatment with CS induced DNA fragmentation indicated by strong red fluorescence staining. In addition, the DNA fragmentation in HCT116 cells by CS treatment occurs in a concentration-dependent fashion.

Observation of CS at 6 µM and 12 µM, induced apoptosis on HCT116 cells by in situ transferase-UTP nick-end labeling (TUNEL) assay. Scale bar: 50 µM (magnification, ×100)
Discussion
Anti-tumor drug discovery and development from plant products plays an important role in cancer chemotherapy. Previous studies have found that some stilbenes, including carboxystilbene, exerted anti-tumor property and therefore could potentially be developed into new chemotherapeutic agents [25]. However, no report has yet addressed the mechanism of apoptosis of carboxamidestilbene (CS) in colon cancer; thus this field remains a promising area for research. In this study, we investigated the cytotoxicity of CS on the colorectal cancer cell line as well its selectivity towards normal colon cells. CS was cytotoxic towards HT29 and HCT116 colon cancer cells in a time- and dose-dependent manner. Interestingly, CS did not show cytotoxicity in normal colon CCD-112 Con cells even up to 72 h of exposure with CS (Table 1). We hypothesize that CS induces cell apoptosis and arrest by mediating multiple apoptotic proteins in HCT116.
Apoptosis and cell cycle arrest are two important mechanisms of anti-cancer drugs. Apoptosis is a highly complex series of molecular events that is energy-dependent, and includes cell shrinkage and DNA fragmentation [16].
There are two pathways involved in apoptosis, namely the extrinsic and intrinsic pathways. The extrinsic signaling pathway involves trans-membrane receptor-mediated interactions whereas the intrinsic pathway involves mitochondria mediated interactions. The ‘death’ domains are key proteins that play an important role in the extrinsic mediated pathway. These domains would transfer the death signal from the cell surface into the nucleus via intracellular signaling pathways [4]. Our mechanism studies show that CS activates the Fas receptor and signals the binding of the adapter protein FADD (Figs. 6, 7). This phenomenon results in the auto-catalytic activation of pro-caspase 8 with the formation of a death-inducing signaling complex (DISC) [19]. Once caspase-8 is activated, it then triggers caspase-3/7, which is an important executioner of the apoptosis process.
Reactive oxygen species (ROS) are produced in the forms of O2−, H2O2, OH, and nitric oxide during the process of active metabolism or stimulation by oncogenes [41, 42]. Various studies have showed that ROS accumulation is very effective in killing cancer cells [14, 29]. Normal cells have a different basal output of ROS compared to cancer cells. This biochemical difference may provide a strong basis for ROS mediated selectivity to kill cancer cells [41]. In this study, we observed that CS treatment enhanced the ROS levels in HCT116 cells and HT-29 but minimally in CCD-112 Con normal colon cells (Fig. 4). Moreover, enhanced Fas activation is also one of the extrinsic pathways of ROS-mediated cell death [6, 24, 42].
All cells undergo mitosis in order to replicate or grow. During this process, the cell duplicates its content and then divides into two daughter cells. The whole process, is governed by several checkpoints through out to ensure the coordinated progression of the cell and to monitor its DNA characteristics as it transits through various stages of the cell cycle [8]. However, there are discrepancy in both assay which we have performed (Annexin V and cell cycle), similar findings were also observed in other publications [21, 38]. However, it is clearly found that in response to external stress, the cell will arrest its cell cycle in order to maintain its genomic integrity. The mechanisms controlling the decision of either to restrain the cell-cycle progression or to induce apoptosis during stress are made through various cell-cycle checkpoints [8]. Various studies have shown that controlling such cell-cycle transition is a good approach in developing chemotherapeutic agents [3, 26, 44]. The analysis of cell cycle over different treatment time points in HCT116 cells revealed a high number of cells (80%) in the G2/M and S phases, as shown in Fig. 3b, whereas the number of cells in the G0/G1 phase decreased in comparison to the untreated cells. The results indicate that CS caused G2/M phase arrest in a time-dependent manner. Proteins p53 and p21waf1/cip1 appear to be crucial in maintaining the G2 checkpoint in human cells [23, 38]. The accumulation of G2/M phase cells post-treatment led us to investigate the expression levels of cell cycle regulator proteins. From our analysis, CS halted the cell cycle progression at the G2/M phase by up-regulating the tumor suppressor proteins, p53 and p21waf1/cip1 (Fig. 7c).
In our findings, we hypothesize that p21waf1/cip1 may cause the release of the pro-apoptotic protein, Bax [1, 31] (Fig. 7c). Several studies have also shown that p21waf1/cip1 plays a pro-apoptotic role, which leads to apoptosis [10, 32]. The disruption of the Bax:Bcl-xL interaction in the mitochondria consequently causes activation of caspase 9 [27, 43]. From our results, Bax was activated, which then led to the activation of caspase 9 (Fig. 5b, c). Elevated levels of caspase 9 from the caspase assay and a similar observation from the western blot analysis both confirm the involvement of caspase 9. Activation of caspase 3 from the activation of caspase 8 and caspase 9 are the hallmarks of apoptosis via both extrinsic and intrinsic pathways. Poly (ADP-ribose) polymerase (PARP) has a particular function in the repair of damaged DNA; the cleavage of PARP is also considered to be one of the hallmarks of apoptosis [17]. The degradation of PARP, impaired DNA repair, and DNA damage/fragmentation were observed in the TUNEL assay (Fig. 8). Collectively, our results support the fact that CS induces apoptosis in HCT116 cells.
In conclusion, the present study demonstrated the anti-tumour effect of CS, which selectively caused cytotoxicity in human colorectal cancer and induces apoptosis. The underlying mechanism of CS is associated with the promotion of ROS levels, activation of the Fas receptor, FADD, caspase 8, caspase 9, caspase 3, cleavage of PARP, disruption of the Bax:Bcl-xL ratio, and cell cycle arrest at G2/M (Fig. 9). The current study highlights the potential of CS as a chemotherapeutic agent for human colon cancer.

CS apoptotic mode of action against HCT116
References
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9(6):400–414
Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y (2004) Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res 24:2783–2840
Alao JP (2007) The regulation of cyclin D1 degredation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 6:24
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281(5381):1305–1308
Chun Fu W, Jing Yu Y, Fang W, Xiao Xiao W (2013) Resveratrol: botanical origin, pharmacological activity and applications. Chin J Nat Med 11:1–15
Denning TL, Takaishi H, Crowe SE, Boldogh I, Jevnikar A, Ernst PB (2002) Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells. Free Radic Biol Med 33:1641–1650
Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW (2015) Targeting mitosis in cancer: emerging strategies. Mol Cell 60(4):524–536
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516
Evan GI, Brown L, Whyte M, Harrington E (1995) Apoptosis and the cell cycle. Curr Opin Cell Biol 7(6):825–834
Gartel AL (2005) The conflicting roles of the cdk inhibitor p21waf1/cip1in apoptosis. Leuk Res 29:1237–1238
Hata T, Yamamoto H, Ngan CY, Koi M, Takagi A, Damdinsuren B, Yasui M, Fujie Y, Matsuzaki T, Hemmi H, Xu X, Kitani K, Seki Y, Takemasa I, Ikeda M, Sekimoto M, Matsuura N, Monden M (2005) Role of p21 waf1/cip1 in effects of oxaliplatin in colorectal cancer cells. Mol Cancer Ther 4(10):1585–1594
Hayward RL, Schornagel QC, Tente R, Macpherson JS, Aird RE, Guichard S, Habtemariam A, Sadler P, Jodrell DI (2005) Investigation of the role of Bax, p21/Waf1 and P53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium (II) organometallic agent, RM175. Cancer Chemother Pharmacol 55(6):577–583
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776
Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W (2000) Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407:390–395
Huerta S, Goulet EJ, Livingston EH (2006) Colon cancer and apoptosis. Am J Surg 191:517–526
IgneyF.H., and Krammer P.H. (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2(4):277–288
Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG (1993) Specific proteolytic cleavage of poly(ADP-ribose) polymerase. Cancer Res 53:3976–3985
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14(22):5579–5588
Leong KH, Looi CY, Loong XM, Cheah FK, Supratman U, Litaudon M, Mustafa MR, Awang K (2016) Cycloart-24-ene-26-ol-3-one, a new cycloartane isolated from leaves of Aglaia exima triggers tumor necrosis factor-receptor 1-mediated caspase-dependent apoptosis in colon cancer cell line. PLoS ONE 11(4)
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W (2012) Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149(6):1269–1283
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5):479–496
Liu S, Bishop WR, Liu M (2003) Differential effects of cell cycle regulatory protein p21WAF1/Cip1 on apoptosis and sensitivity to cancer chemotherapy. Drug Resist Update 6:183–195
Medan D, Wang L, Toledo D, Lu B, Stehlik C, Jiang BH, Shi X, Rojanasakul Y (2005) Regulation of Fas (CD95)-induced apoptotic and necrotic cell death by reactive oxygen species in macrophages. J Cell Physiol 203:78–84
Mohamad NA, Mohd FMD, Chin HK, Munirah S, Ang KP, Kartini A, Mohd AN, Noel FT, Mohamad K, Leong KH, Awang K (2013) Design, synthesis and cytotoxic evaluation of o-carboxamidostilbene analogues. Int J Mol Sci 14:23369–23389
Molinari M (2000) Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif 33:261–274
Naseri MH, Mahdavi M, Davoodi J, Tackallou SH, Goudarzvan M, Neishabouri SH (2015) Upregulation of Bax and downregulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int 15:55
Nuñez G, Benedict MA, Hu Y, Inohara N (1998) Caspases: the protease of the apoptotic pathway. Oncogene 17:3237–3245
Pelicano H, Carney D, Huang P (2004) ROS stress in cancer cells and therapeutic implications. Drug Resist Updates 7:97–110
PientenpolJ. A., and Stewart Z.A. (2002) Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology 181–182:475–481
Pucci B, Kasten M, Giordano A (2000) Cell cycle and apoptosis. Neoplasia 2(4):291–299
Qiu P, Guan H, Dong P, Li S, Ho C, Pan M, McClements D, Xiao H (2010) The p53-, Bax- and p21-dependent inhibition of colon cancer cell growth by 5-hydroxy polymethoxyflavones. Mol Nutr Food Res 55(4):613–622
Savouret JF, Quesne M (2002) Resveratrol and cancer: a review. Biomed Pharmacother 56:84–87
Schatzkin A, Kelloff G (1995) Chemo- and dietary prevention of colorectal cancer. Eur J Cancer 31A:1198–1204
Shapiro GI, Harper JW (1999) Anticancer drug targets: cell cycle and checkpoint control. J Clin Investig 104(12):1645–1653
Shi Y (2004) Caspase activation, inhibition and reactivation: a mechanistic view. Protein Sci 12(8):1979–1987
Taylor RC, Cullen SP, Martin SJ (2008) Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9:231–241
Taylor WR, Stark GR (2001) Regulation of the G2/M transition by p53. Oncogene 20:1803–1815
Thakur VS, Gupta K, Gupta S (2011) Green tea polyphenols causes cell cycle arrest and apoptosis in prostate cancer cells by suppressing class I histone deacetylases. Carcinogenesis 33(2):377–384
Torre L, Siegel R, Jemal A (2015) American cancer society. Global cancer facts & figs, 3rd edn. American Cancer Society, Atlanta
Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethylisothiocyanate. Cancer Cell 10:241–252
Uchikura K, Wada T, Hoshino S, Nagakawa Y, Aiko T, Bulkley GB, Klein AS, Sun Z (2004) Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. Am J Physiol Gastrointest Liver Physiol 287:G620–G626
Waris G, Ahsan H (2006) Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog 5:14
Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408:433–439
Acknowledgements
We are grateful to the University of Malaya Postgraduate Research Grant (PPP) (PG077-2014A) and the Ministry of Higher Education Fundamental Research Grant (FP024-2014A) for the financial supports.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Cheah, F.K., Leong, K.H., Thomas, N.F. et al. Resveratrol analogue, (E)-N-(2-(4-methoxystyryl) phenyl) furan-2-carboxamide induces G2/M cell cycle arrest through the activation of p53–p21CIP1/WAF1 in human colorectal HCT116 cells. Apoptosis 23, 329–342 (2018). https://doi.org/10.1007/s10495-018-1457-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-018-1457-8