
Abstract
The concept to fight against tumour resistance is to use chemosensitizers that selectively sensitize tumour cells to chemotherapeutic drugs without affecting normal tissue. In this study, the chemosensitizing potential of a novel benzoxazine derivative in combination with Doxorubicin, a DNA damaging chemotherapeutic drug was evaluated. The results of this study showed that the compound LTUR6 is a potent chemosensitizer of Doxorubicin in colon cancer cell lines, HCT116 and HT29. It was also observed that LTUR6 delayed the resolution of Doxorubicin-induced γH2AX, a specific marker of unrepaired DNA DSB, and prolonged cell cycle arrest in both cell lines. This eventually led to DNA fragmentation, caspase activation and ultimately apoptosis in LTUR6 treated cell lines. Results of western blot analysis revealed that LTUR6 significantly inhibited the phosphorylation of DSB repair enzyme AKT, in response to Doxorubicin-induced DSB. We propose that the chemosensitization observed following inhibition of PI3K is likely due to the involvement of a number of downstream targets of AKT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Colorectal cancer is the second most common cause of cancer-related mortality in Western countries [1]. Around 30% of the patients have advanced disease at presentation, either locally or at distant sites; where chemotherapy is the only therapeutic option. However, at times, the inherent resistance of metastatic colon cancer to many currently established treatment regimens remains a major hindrance in cancer therapy. Doxorubicin is widely used in the treatment of colon cancer [2,3,4]. It is a topoisomerase II inhibitor and inhibition of this enzyme can result in DNA-double strand breaks (DSBs) [5]. However, cells are programmed with various DNA-DSB repair pathways such as Non-Homologous end-joining (NHEJ) and Homologous-recombination (HR) followed by a transient halt in the cell cycle [6]. These DSB repair mechanisms are one of the main causes of cancer cell resistance and failure of chemotherapy [7]. In response to DSBs, AKT is activated and its activity is modulated downstream by phosphatidylinositol 3 kinase (PI3K) through a multistep process. PI3Ks are lipid kinases that have been implicated in many physiological processes such as cell cycle regulation, DNA repair, apoptosis, senescence, angiogenesis and cellular metabolism and pathological disorders such as cancer [8]. PI3K is involved in the repair of DSBs by recruitment of resection factors, such as RPA, Brca1, and Rad51, to sites of damage [9, 10]. Consequently, inhibition of this enzyme will impede the repair of cancer cells prolonging cell cycle arrest leading to apoptosis.
The PI3K-AKT signalling pathway regulates cell growth and survival through various mechanisms. Deregulation of the PI3K signalling pathway has been identified in approximately one-third of all cancers [11, 12]. The role of PI3K isoforms in the context of tumorigenesis appears to be both coinciding and distinctive. PI3Kα has been involved in cancer cell proliferation and tumour progression [13]. PI3Kβ has been associated with transformation induced by the inactivation of PTEN, both in vivo and in vitro [14]. High expression of PI3Kγ has been observed in chronic myeloid leukaemia [15]. Overexpression of PI3Kδ has been observed in acute myeloblastic leukaemia and some studies have demonstrated its role as an oncoprotein [16,17,18]. PI3Kδ has also been implicated in tumour angiogenesis, particularly in the conditions of repair following damage of tumour blood vessels by radiation [19]. Although expression of the γ and δ isoforms is normally restricted to leukocytes, increased p110δ has been identified in some colon malignancies [20]. Recently, there has been an upsurge in the development of isoform-specific PI3K inhibitors that sensitize cancer cells to chemo and radiotherapy. However, very few have proved promising enough to enter clinical trials. This might be due to the undesirable pharmacokinetic profile of the drugs and partly due to the lack of correlation between in vitro studies and early clinical trials. Several lines of evidence suggest redundancy in PI3K isoform signalling and additionally deregulated PI3K pathway signalling has been shown to confer resistance to conventional therapies and radiation [21,22,23].
Several studies have reported cardiotoxicity following treatment with Doxorubicin and have worked out to optimize treatment regimens including anthracyclines such as Doxorubicin [24, 25]. These include dose optimization, combination therapy, adjuvant therapy, use of iron-chelating agents and modified drug formulations [26, 27]. Despite significant research being devoted to circumvent cardiotoxicity without compromising its antitumor efficacy, to date none of the current approaches have proven to be successful [28]. This draws attention to the potential role of chemosensitizers as an adjunct to Doxorubicin. Chemosensitizers can selectively enhance the cytotoxic effects of anti-cancer drugs without exerting toxic effects on normal tissues. Thus at a minimum possible therapeutic dose, maximum antitumor efficacy can be obtained without dose escalation and hence dose-dependent toxic effects can be minimized. Here, the role of a novel benzoxazine derivative, LTUR6 a PI3K inhibitor, as a potential chemosensitizer in combination with Doxorubicin, was investigated.
Materials and methods
Cell lines and cell culture
The human colon tumours cells (HT-29 and HCT-116) were used to study the effects of compounds on chemosensitivity. Both the cell lines were obtained from the American Type Culture Collection (ATCC, Maryland, USA). Cells were grown in DMEM media (Thermo Scientific HyClone) containing 10% FBS (In Vitro Technologies, Auckland, New Zealand), 4.5 g glucose/L, 4 mM L-glutamine and sodium pyruvate and maintained in a humidified atmosphere at 37 °C with 5% CO2.
Doxorubicin and sensitizing compounds
Novel benzoxazine derivatives were synthesized in the Organic Chemistry Laboratory, La Trobe University, Bendigo. Selection of the test inhibitor was based on its IC50 against PI3K isotypes and DNA-PK as assayed by Reaction Biology Corporation USA (Table 1). The compound LTUR6 was selected based on the expression levels of PI3Kδ in the cell lines used (HT-29 and HCT116) for analysis. HT-29 expresses low levels of normal PI3Kδ but HCT-116 has a deletion of exon 16 which should mean a loss of function [Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations]. A known PI3K inhibitor (LY294002) was used for comparisons [22, 29, 30]. LY294002 was obtained from Cell Signalling Technology (Danvers, MA). Stock solutions of 1 mM were prepared in DMSO and stored at −20 °C and further diluted in culture medium to obtain the required concentration. The percentage of DMSO was kept below 0.5% to avoid toxicity [31]. Doxorubicin, topoisomerase II inhibitor, was dissolved in Milli-Q H2O and stored at −20 °C. A stock solution of 1 mM was prepared from which further dilutions were made in media.
Colony formation assay
Colony formation assay was performed on both cell lines HT-29 and HCT-116. Cells were seeded homogenously over the entire surface area at a density of 200 cells per well in a 6-well plate and left overnight to attach. The following day, cells were treated with control, Doxorubicin, compounds (LTUR6 and LY294002) alone or in combination with Doxorubicin at desired concentrations and cultured for 2 days. Media was aspirated and cells were washed thoroughly with PBS to remove any excess media containing drug. Fresh culture media with no drug was added and replaced every 2 days. After 9 days, cells were washed again with PBS and fixed using methanol:acetic acid 3:1 solution. After fixing, cells were stained using crystal violet for visualization. Colonies were counted using a Syngene automated colony counter. Colonies were defined as a cluster of at least 50 cells.
Gamma H2AX assay for DNA damage
This assay was performed as described by MacPhail et al. [32].HT-29 and HCT-116 cells were plated in a six well plate at a seeding density of 1.5–2.0 × 105 cells/well and left overnight to adhere. The following day, the seeded wells were treated with control, Doxorubicin, compounds (LTUR6 and LY294002) alone or in combination with Doxorubicin at desired concentrations for 4 or 4 h treatment with 24 h recovery. Cells were then fixed using 70% ethanol. Before fixing, the media was collected and stored in an Eppendorf tube (to avoid the loss of any suspended cells); attached cells were then washed with PBS to remove excess media. Cells were detached using 1 mL Trypsin-Versene and incubated at 37 °C for 1–2 min. This media was collected and added to the previously collected media, and centrifuged at 200 g for 5 min at 6 °C. Following centrifugation, the supernatant was removed and 500 μL of 70% ethanol was added into the cell pellet and stored at 20 °C for 20 min. Pellets were washed twice in Tris buffered saline and once in TFX and allowed to rehydrate on ice for 10 min. The cell suspension was centrifuged at 200×g for 5 min at 4 °C, and resuspend in 100 μL rabbit polyclonal anti-p-histone H2AX diluted at 1:500 in TFX. The cell suspension was incubated at room temperature for 2 h under constant agitation. After 2 h, the cells were washed with TFX and resuspended in 100 μL secondary antibody, goat anti-rabbit IgG Alexa fluor 488 fragments diluted at 1:200 in TFX and incubated at room temperature for 1 h under agitation, protected from light. The cells were washed in TFX and resuspend in 300 μL of propidium iodide 5 μg/mL. Fluorescence was read using a flow cytometry analysis on the Accuri C6 Flow cytometer (BD Biosciences, San Jose) and data analysed using CFlow software. Log fluorescence against cell count was plotted.
Cell cycle analysis
Cell cycle progression was analysed to determine involvement of checkpoint activation or cell cycle arrest. HCT116 and HT29 cells were plated in a six well plate a seeding density of 2 × 105cells per well and treated with desired concentrations of test inhibitors (LTUR6 and LY294002) for 24 h. Following this, media from the cell culture was aspirated and retained and the cells were detached using 1 mL trypsin incubated at 37 °C for 1 to 2 min. The trypsin with detached cells was then added to the previously collected media, and centrifuged at 200×g for 5 min at 6 °C. Following centrifugation, the supernatant was removed and 500 μL of 70% ethanol was added and stored at 20 °C for 20 min. The cell suspension was centrifuged again for pellet formation at 200×g for 5 min at 4 °C. The supernatant was removed carefully without disturbing the pellet. The cells were washed with PBS and resuspended in staining reagent (mixture of 25 μg/mL of propidium iodide and 100 μg/mL RNaseA in PBS) and incubated at 37 °C for 30 min. Cells were then stored on ice and analysed using flow cytometry (Accuri C6 Flow cytometer, BD Biosciences, San Jose). A total of 10,000 events were recorded for each sample.
Apostat assay
This assay was used to measure activation of caspases using FITC-conjugated, pan-caspase inhibitor (ApoStat; R&D Systems, Minneapolis, MN). The assay was performed according to the manufacturer’s instructions. Cells were plated at a seeding density of 106 cells per well of a 24 well plate. They were treated with control, Doxorubicin, compounds alone (LTUR6 and LY294002) or in combination with Doxorubicin at desired concentrations for 4 h. Following this the media was aspirated and replaced with fresh media containing inhibitors alone in combination treated wells and DMSO in Doxorubicin treated wells and incubated for 24 h. Cells were then stained during the last 30 min of the apoptosis induction period. For this, 10 mL of ApoStat per 1 mL culture volume was added and incubated at 37 °C. After the staining period, cells were harvested into 5 mL tubes, centrifuged at 500×g for 5 min and washed once with 4 mL of PBS to remove unbound reagent. Cells were resuspended in 500 µL of PBS for flow cytometric analysis. Results are presented as % of cells containing active caspases.
Western blotting
For preparation of lysates, cells were exposed to control, Doxorubicin, compounds alone or combination of compound and Doxorubicin, for 30 min. Cells were harvested, and washed twice with ice-cold PBS containing 1 mM PMSF (phenylmethylsulphonyl fluoride) and resuspended in 100 μL of ice cold lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 1× protease inhibitor cocktail (Roche), 1 mM PMSF and 10 μg/mL aprotinin, in distilled water). Samples were centrifuged (14,000 rpm, 25 min at 4 °C) and the supernatants were transferred into pre-chilled eppendorf tubes after 20 min incubation on ice and were placed on ice pending analysis. The protein concentration of each sample was determined using the Bio-Rad DC protein assay kit (Bio-Rad) according to the manufacturer’s instructions. Typically 50 μg of protein per sample was denatured via addition of 1× SDS loading buffer (abcam, ab119196) in the presence of 100 mM DTT (abcam) and heated for 10 min at 75 °C. Samples were loaded onto 4 or 10% gels (abcam) based on the molecular weight of the protein of interest. Samples were run in the presence of SDS running buffer (abcam, ab119195), typically at 100 V for approximately 90 min to separate proteins. Low molecular weight proteins were separated using 10% gels and high molecular weight proteins separated using 4% gels. Samples were run until the dye front reached the bottom of the gel. After running, the gels were removed from casing and used for Western transfer.
Western transfer for proteins was carried out using a Mini trans-Blot apparatus from Bio-Rad. Gels were equilibrated for 10 min in 1× transfer buffer (48 mM Tris-HCl (pH 7.5), 38.5 mM glycine, 0.037% SDS and 20% (v/v) methanol). Polyvinylidene fluoride (PVDF) membranes were activated in 100% methanol and equilibrated in 1× transfer buffer for 10 min. Whatman filter paper (two for each gel) was soaked in 1× transfer buffer along with transfer sponges. The transfer cassette was assembled according to manufactures instruction. The tank was filled with transfer buffer and transfer of protein was done by applying an electric current (50 V, 250 mA) for 3 h in a 4 °C cold room. Following transfer, the PVDF was carefully removed and probed with antibodies by blocking the PVDF membranes in 10% (w/v) bovine serum albumin in TBS-T (Tris-buffered saline containing 0.1% (v/v) Tween-20) for 2 h. The membranes were then washed three times; 5 min per wash, in TBS. Membranes were exposed to primary antibodies at the appropriate dilution (1/5000) overnight at 4 °C. The primary antibody was removed by washing three times, 5 min per wash in TBS-T. Membranes were then exposed to secondary antibodies (abcam ab97200, HRP conjugated goat anti-rabbit IgG, 1 in 10,000 dilution in TBS-T) for 2 h at room temperature. The secondary antibody was removed by washing three times in TBS-T. After washing, the membranes were exposed to ChemiFast Chemiluminescence substrate (Syngene, USA, CH-FAST/20). Images were captured using Syngene G-BOX (G: BOX-CHEMI-XL1.4, USA). For quantifying the density of proteins, the integrated optical density (IOD) of each band was read at increasing exposures using a Bio-Rad Chemi Doc station. IOD values were corrected for background signal. Values were normalized to those of the controls and the protein expression levels were then quantified.
Results
Chemosensitization and toxicity profile of LTUR6 using clonogenic assay of HCT116 and HT29
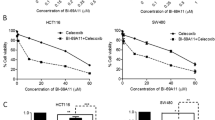
After 7 days in the presence 0.1 μM LTUR6 or 1.0 μM LY294002 no inhibition of colony formation in HCT116 (Fig. 1a) or HT29 (Fig. 1b) was seen. Doxorubicin alone was seen to significantly inhibit colony formation at 2.5 µM (p < 0.05) which was further enhanced significantly (p < 0.05) in combination with LTUR6 or LY294002.

Analysis of clonogenic assay. HCT116 cells (a) and HT29 (b) cells were cultured in the presence of media alone (control), Doxorubicin (Dox, 2.5 μM), LTUR6 (0.1 μM), and LY294002 (1 μM) results shown after 7 days. Graph indicates average of three replicates. Error bars represent the Standard Error. Statistical significance was determined by one way ANOVA. *Indicates groups significantly different to Doxorubicin alone, (p < 0.05)
Induction of DSBs and their repair in HCT116 and HT29 cells
After 4 h in culture media alone, analyses of DSBs in HCT116 showed basal levels, or minimal levels in the in vitro culture environment (Fig. 2a). When cells were exposed to Doxorubicin, the levels of DSBs increased significantly compared to control. When the HCT116 cells were exposed to either LTUR6 or LY294002 alone no significant differences were noted in the amount of DSB detected when compared to the control. When Doxorubicin was combined with LTUR6, there was a significant (p < 0.05) increase in the number of DSB detected after 4 h when compared to Doxorubicin alone. LY294002, when combined with Doxorubicin, significantly increased DSB compared to Doxorubicin alone, but this was not as great an increase as that seen with the combination of Doxorubicin and LTUR6. A similar trend was observed in HT29 when exposed to Doxorubicin in combination with LTUR6 (Fig. 2b).

Representative histograms and bar graphs data depicting DSBs induced in a HCT116 cells and b HT29 at 4 h. Cells were exposed to 0.1 μM LTUR6 or 1 μM LY294002 alone or in combination with 5 μM Doxorubicin. A shift towards the right indicates increase in fluorescence which corresponds to an increase in γ-H2AX
Next HCT116 and HT29 cells were exposed to Doxorubicin with or without compounds for 4 h and were then allowed to recover in the presence of compounds for 24 h (Fig. 3a, b). A similar trend was observed, as that seen after 4 h. However, in Doxorubicin treated cells, the levels of DSB increased significantly (p < 0.05) compared to control even after 24 h recovery though, by comparison with the 4 h treatment, this was not as great an increase over control. When Doxorubicin was combined with LTUR6, there was a significant increase in the number of DSB detected even after the 24 h recovery time, when compared to Doxorubicin alone. LY294002, when combined with Doxorubicin, showed no significant effect over Doxorubicin alone.

Representative histograms and bar graphs depicting DSBs induced in a HCT116 cells and b HT29 after 4 h Doxorubicin treatment and 24 h recovery period. Cells were exposed to 0.1 μM LTUR6 or 1 μM LY294002 alone or in combination with 5 μM Doxorubicin. A shift towards the right indicates increase in fluorescence which corresponds to an increase in γ-H2AX
Cell cycle analysis
When HCT116 cells were exposed to Doxorubicin, a significant increase in cells in G2/M-phase was observed when compared to control, (p < 0.05) with a concurrent decrease in G0/G1-phase (Fig. 4a, c). Doxorubicin induced G2/M cell cycle arrest was further enhanced in combination with LTUR6. Doxorubicin treatment in combination with LY294002 showed a similar though lesser build-up of cells in G2/M to the Doxorubicin and LTUR6 combination but also showed a build-up of cells in S phase which was significant compared to control, (p < 0.05).

Representative graphs indicating the cell cycle profile of HCT116 (a, c) and HT-29 (b, d) cells after exposed to 0.1 μM LTUR6 and 1 μM LY294002 alone or in combination with 5 μM Doxorubicin after 24 h recovery period. Red bars show the percentage of cells in each phase of the cell cycle. (Color figure online)
When HT29 cells were exposed to Doxorubicin, the results indicate a significant (p < 0.05) increase in cells in S-phase as compared to control, with a concurrent decrease in G2/M-phase (Fig. 4b, d). In combination with test compounds LTUR6 and LY294002, a shift in the cell cycle profile was observed indicating increased cells in G2/M paralleled with a decrease in S and G0/G1-phase. This effect was again stronger with LTUR6 than with LY294002.
Apoptosis induced in cells undergoing cell cycle arrest
Apoptosis was measured by apostat assay following 4 h Doxorubicin exposure in HCT116 (Fig. 5a) and HT29 (Fig. 5b) cells. It was noted that a significant (p < 0.05) level of apoptosis was observed in Doxorubicin treated cells which was further enhanced in combination with 0.1 μM LTUR6. This increase in apoptosis was very evident in the case of LTUR6 (p < 0.05) compared to LY294002 as shown by Apostat assay.

Representative histogram and bar graphs indicating the activation of caspases in HCT116 (a) and HT29 (b) after 4 h Doxorubicin treatment and 24 h recovery time. Cells were exposure to 0.1 μM LTUR6 or 1 μM LY294002 alone or in combination with 5 μM Doxorubicin. Marker indicates population of cells expressing activated caspases. The data obtained represent the average of three independent experiments
Phosphorylation status of AKT and DNA-PK during chemosensitization with LTUR6 and Doxorubicin
The results of Western blotting in HCT116 and HT29 confirmed that LTUR6 in combination with Doxorubicin inhibited AKT phosphorylation at Ser473 and Thr308. LY294002 failed to inhibit AKT phosphorylation (Fig. 6a, b). However, the results of Western blotting in HCT116 and HT29 confirmed that LTUR6 in combination with Doxorubicin failed to inhibit DNA-PK phosphorylation at Ser2056 and Thr2609 (Fig. 7a, b).

a Phosphorylation of AKT1 at Ser473 and Thr308 in HCT116 cells was analysed by Western blotting. Cells were treated with DMSO (control), Doxorubicin (5 μM) alone or in combination with test compounds (1 μM LY294002and 0.1 μM LTUR6) for 4 h. Proteins levels quantified by measuring the pixel densities were plotted in the form of a bar diagram. b Phosphorylation of AKT1 at Ser473 and Thr308 in HT29 cells was analysed by Western blotting. Cells were treated with DMSO (control), Doxorubicin (5 μM) alone or in combination with test compounds (1 μM LY294002 and 0.1 μM LTUR6) for 4 h. Proteins levels quantified by measuring the pixel densities were plotted in the form of a bar diagram

a Phosphorylation of DNA-PKcs at Ser2056 and Thr2609 in HCT116 cells was analysed by Western blotting. Cells were treated with DMSO (control), Doxorubicin (5 μM) alone or in combination with test compounds (1 μM NU7026 and 0.1 μM LTUR6) for 4 h. b Phosphorylation of DNA-PKcs at Ser2056 and Thr2609 in HT29 cells was analysed by Western blotting. Cells were treated with DMSO (control), Doxorubicin (5 μM) alone or in combination with test compounds (1 μM NU7026 and 0.1 μM LTUR6) for 4 h
Discussion
In this study we effectively demonstrated the effect of a novel benzoxazine, LTUR6, to sensitize human colon cancer cell lines to the effects of Doxorubicin. LTUR6, a PI3Kδ specific inhibitor exhibited no toxicity of its own. A previous study conducted on Ewing’s sarcoma cell line, TC-71, reported that the combined treatment with the broad specificity PI3K inhibitor LY294002 (10 μM) and Doxorubicin (3–30 μg/mL) significantly enhanced cell growth inhibition as determined by clonogenic assay [33]. Another study done on SK-OV-3 (ovarian carcinoma), MDA-MB-468 (breast carcinoma), and A549 (lung carcinoma) cell lines demonstrated enhancement of Doxorubicin-induced growth inhibition by LY294002 at 10 μM [34]. LTUR6 at a concentration of 0.1 μM showed greater potency compared to LY294002, sensitizing both cell lines to Doxorubicin.
Following DNA damage, the H2AX protein at the site of damage is phosphorylated by kinases including ATM and is then referred to as γ-H2AX. Following DNA repair, γ-H2AX gets dephosphorylated by a phosphatase, PP2A, responsible for the regulation of γ-H2AX levels in human cells and consequently detection of γ-H2AX can be used in the assessment of DNA repair kinetics [35]. DNA DSBs induced by Doxorubicin were markedly increased in the presence of LTUR6 in both cell lines after 4 h exposure. DNA-PK, ATM and ATR contribute to the activation of H2AX depending on a number of factors such as the cell cycle stage, the type of DNA DSB and the relative expression levels of individual PIKKs, a process which is not yet fully understood [36,37,38]. Therefore, a known specific inhibitor of PI3K (LY294002) was included in the assays to analyse the relative roles of this protein in the repair of DSBs. A study done on SV40-transformed human fibroblasts derived from skin biopsies showed a synergistic effect between Doxorubicin and 10 μM LY294002 in inducing DSBs in DNA repair deficient cell lines [39]. This finding is consistent with our data where both Doxorubicin-treated cell lines accumulated slightly higher levels of DSBs in the presence of LTUR6 and LY294002 at much lower concentrations.
To understand whether the effect observed was on DNA damage induction alone or/and repair, we examined the DNA repair kinetics in HCT116 and HT-29 cell lines after a 24 h recovery period following an initial exposure to Doxorubicin with/without test compounds. As indicated by Figs. 3 and 4, both the cell lines could complete DSB repair in the LY294002 treated groups by 24 h after DSB induction. However, LTUR6 significantly impeded the repair of DNA DSBs induced by Doxorubicin (Fig. 4b, d, p < 0.05). Even after 24 h, the level of DSBs in LTUR6 treated groups was significantly higher indicating incomplete DSB repair, (p < 0.05). These data suggest that LTUR6 might retard the repair mechanism of Doxorubicin-induced DNA damage.
Inducing DNA damage triggers DNA damage repair (DDR) which begins with cell cycle arrest to facilitate repair mechanisms. In HT-29 cells, exposure to Doxorubicin alone caused a profound increase of cells in S-phase. This is consistent with the previous observation in colon cancer cells where p53-mutant SW620 cells showed a marked S/G2 accumulation in response to Doxorubicin [40]. Strikingly, concomitant treatment with Doxorubicin and LY294002 converted the S-phase arrest into G2/M arrest. This profound G2/M-block in response to combination treatment was in line with a study demonstrating co-exposure with andrographolide and LY294002 enhanced G2/M arrest in human glioblastoma U251 and U87 cells [41]. Our compound LTUR6 also showed a similar though even more pronounced G2/M arrest to that of LY294002. In HCT-116 cells, we observed that incubation of these cells with Doxorubicin alone resulted in accumulation of cells in G2/M phase and a concurrent decrease in the fraction of cells in S and G0/G1 phases. The observed effect of Doxorubicin is in good agreement with a study conducted by Potter et al. reporting that Doxorubicin-induced DNA damage was predominantly in the G2 phase of the cell-cycle [42]. Interestingly, LY294002 demonstrated a shift in the cell cycle arrest from G2/M-block to S-block in presence of Doxorubicin. This might possibly be due to the broad specificity of this compound against various PI3K enzymes. LTUR6 on the other hand inhibited the transition of cells from G2/M to G0/G1 phase. This finding is in line with a study documenting that silibinin, a derivative of milk thistle, induced G2–M arrest in combination with Doxorubicin [43]. To recapitulate, Doxorubicin induced cell cycle arrest was markedly enhanced in the presence of LTUR6 and this was greater than that observed with LY294002. Hence, it can be concluded that increased DSBs in presence of this benzoxazine has instigated DDR by initiating cell cycle arrest which is evidenced by the results discussed above.
Following failure of DSB repair and cell cycle arrest, cell killing by apoptosis in response to chemotherapeutic agents is considered to be a common pathway [44]. The apostat assay measures total activation of caspases, an essential event in apoptosis [45]. Doxorubicin alone caused an increase in apoptosis compared to control in both the cell lines tested. This apoptosis level was further enhanced significantly in the presence of LTUR6 in both the cell lines. Cells treated with LY294002 in combination with Doxorubicin showed no significant difference in apoptosis. This was in contrast to a study carried out on the human leukaemic cell line HL60, where a 30-fold higher concentration of LY294002 (30 μM) in the presence of varying concentrations of Doxorubicin induced apoptosis [46]. The fact that LY294002 is considerably less potent in enhancing apoptosis after treatments known to induce DSB’s than LTUR6 could be due the selective inhibition of LTUR6 against PI3Kδ. The relative expression of PI3K enzymes in these cell lines and the difference in the IC50 values between LTUR6 and LY294002 may account for the varying degree of DSB repair inhibition and the varying degree of apoptosis observed. This remains to be investigated.
As AKT is a downstream target of both PI3K and DNA-PK and is a key signalling molecule in pathways leading to apoptosis and cell cycle arrest, we assessed the phosphorylation status of AKT in both the cell lines following exposure to LTUR6 in combination with Doxorubicin [47]. LTUR6 in combination with doxorubicin inhibited the phosphorylation of AKT at Thr308 and Ser473. The effect observed was similar in both HCT116 and HT29 cell lines. Doxorubicin alone induced AKT phosphorylation on both Thr308 and Ser473 which agreed with a previous study using similar concentration of Doxorubicin, 5 μM [48]. Interestingly, in combination with LY294002, the phosphorylation status of AKT appeared to be the same as in the Doxorubicin alone treated group. A previous study showed a dose and time dependent increase in the phosphorylation of AKT in the presence of Doxorubicin in several breast cancer cell lines. This effect was abolished when cells were concomitantly exposed to LY294002, though at a much higher concentration of 10 µM, indicating that the doxorubicin-induced phosphorylation and activation of AKT were mediated through a PI3-K dependent pathway [49]. As AKT has been shown to be an important downstream target of DNA-PK we have analysed the possibility DNA-PK inhibition that would subsequently inhibited the phosphorylation of AKT. However, LTUR6 failed to inhibit the phosphorylation of DNA-PK at Ser2056 and Thr2609 residues in both cell lines.
The overall aim of this study was to identify the molecular pathways that were altered to enhance Doxorubicin-mediated cytotoxicity in presence of the novel benzoxazine derivative, LTUR6. From the results it can be seen that protein kinases such as AKT plays key role in response to DNA DSBs repair machinery, and their inhibition in presence of LTUR6 enhanced the sensitivity of Doxorubicin. Further investigation is required to determine the involvement of a number of downstream targets of AKT including RSK1/2/3, mTOR, p38, PRAS40, HSP27, EGFR, FAK, STAT and GSK-α/β that regulate cell cycle arrest, DNA repair and apoptosis. As such, knowledge about the molecular determinants of LTUR6-mediated chemosensitization will aid in developing more potent drugs that will limit concurrent damage to normal tissues and improve the quality of life of cancer patients.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12):2893–2917. doi:10.1002/ijc.25516
Lupertz R, Watjen W, Kahl R, Chovolou Y (2010) Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 271(3):115–121. doi:10.1016/j.tox.2010.03.012
Watson JL, Hill R, Lee PW, Giacomantonio CA, Hoskin DW (2008) Curcumin induces apoptosis in HCT-116 human colon cancer cells in a p21-independent manner. Exp Mol Pathol 84(3):230–233. doi:10.1016/j.yexmp.2008.02.002
Ravizza R, Gariboldi MB, Passarelli L, Monti E (2004) Role of the p53/p21 system in the response of human colon carcinoma cells to Doxorubicin. BMC Cancer 4:92. doi:10.1186/1471-2407-4-92
Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9(5):338–350. doi:10.1038/nrc2607
Bartlett EJ, Brissett NC, Plocinski P, Carlberg T, Doherty AJ (2016) Molecular basis for DNA strand displacement by NHEJ repair polymerases. Nucleic Acids Res 44(5):2173–2186. doi:10.1093/nar/gkv965
Hao C, Shao R, Raju U, Fang B, Swisher SG, Pataer A (2016) Accumulation of RNA-dependent protein kinase (PKR) in the nuclei of lung cancer cells mediates radiation resistance. Oncotarget 7(25):38235–38242. doi:10.18632/oncotarget.9428
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296(5573):5573. doi:10.1126/science0.1655
Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E (2014) PI3K/AKT signaling pathway and cancer: an updated review. Ann Med 46(6):372–383. doi:10.3109/07853890.2014.912836
Ocana A, Vera-Badillo F, Al-Mubarak M, Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD, Seruga B, Pandiella A, Amir E (2014) Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PLoS ONE 9(4):e95219. doi:10.1371/journal.pone.0095219
Shaw RJ, Cantley LC (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441(7092):424–430. doi:10.1038/nature04869
Arteaga CL (2010) Clinical development of phosphatidylinositol-3 kinase pathway inhibitors. Curr Top Microbiol Immunol 347:189–208. doi:10.1007/82_2010_54
Yuan TL, Choi HS, Matsui A, Benes C, Lifshits E, Luo J, Frangioni JV, Cantley LC (2008) Class 1 A PI3K regulates vessel integrity during development and tumorigenesis. Proc Natl Acad Sci USA 105(28):9739–9744. doi:10.1073/pnas.0804123105
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM (2005) The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci USA 102(51):18443–18448. doi:10.1073/pnas.0508988102
Hickey FB, Cotter TG (2006) BCR-ABL regulates phosphatidylinositol 3-kinase-p110gamma transcription and activation and is required for proliferation and drug resistance. J Biol Chem 281(5):2441–2450. doi:10.1074/jbc.M511173200
Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, Hunault-Berger M, Berthou C, Villemagne B, Jourdan E, Audhuy B, Solary E, Witz B, Harousseau JL, Himberlin C, Lamy T, Lioure B, Cahn JY, Dreyfus F, Mayeux P, Lacombe C, Bouscary D (2005) Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 106(3):1063–1066. doi:10.1182/blood-2004-08-3225
Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE (2003) Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol 170(5):2647–2654
Sadhu C, Dick K, Tino WT, Staunton DE (2003) Selective role of PI3K delta in neutrophil inflammatory responses. Biochem Biophys Res Commun 308(4):764–769
Geng L, Tan J, Himmelfarb E, Schueneman A, Niermann K, Brousal J, Fu A, Cuneo K, Kesicki EA, Treiberg J, Hayflick JS, Hallahan DE (2004) A specific antagonist of the p110delta catalytic component of phosphatidylinositol 3′-kinase, IC486068, enhances radiation-induced tumor vascular destruction. Cancer Res 64(14):4893–4899. doi:10.1158/0008-5472.CAN-03-3955
Kang S, Denley A, Vanhaesebroeck B, Vogt PK (2006) Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci USA 103(5):1289–1294. doi:10.1073/pnas.0510772103
Burris HA (2013) Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol 71(4):829–842. doi:10.1007/s00280-012-2043-3
Chaussade C, Rewcastle GW, Kendall JD, Denny WA, Cho K, Gronning LM, Chong ML, Anagnostou SH, Jackson SP, Daniele N, Shepherd PR (2007) Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem J 404(3):449–458. doi:10.1042/BJ20070003
Miller TW, Balko JM, Arteaga CL (2011) Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol 29(33):4452–4461. doi:10.1200/JCO.2010.34.4879
Alsaad AM, Zordoky BN, El-Sherbeni AA, El-Kadi AO (2012) Chronic doxorubicin cardiotoxicity modulates cardiac cytochrome P450-mediated arachidonic acid metabolism in rats. Drug Metab Dispos 40 (11):2126–2135. doi:10.1124/dmd.112.046631
An L, Hu XW, Zhang S, Hu X, Song Z, Naz A, Zi Z, Wu J, Li C, Zou Y, He L, Zhu H (2017) UVRAG deficiency exacerbates doxorubicin-induced cardiotoxicity. Sci Rep 7:43251. doi:10.1038/srep43251
Lao J, Madani J, Puertolas T, Alvarez M, Hernandez A, Pazo-Cid R, Artal A, Anton Torres A (2013) Liposomal Doxorubicin in the treatment of breast cancer patients: a review. J Drug Deliv 2013:456409. doi:10.1155/2013/456409
Leonard RC, Williams S, Tulpule A, Levine AM, Oliveros S (2009) Improving the therapeutic index of anthracycline chemotherapy: focus on liposomal doxorubicin (Myocet). Breast 18(4):218–224. doi:10.1016/j.breast.2009.05.004
Tacar O, Sriamornsak P, Dass CR (2013) Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 65(2):157–170. doi:10.1111/j.2042-7158.2012.01567.x
Izzard RA, Jackson SP, Smith GC (1999) Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res 59(11):2581–2586
Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW (2003) Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res 63(18):6008–6015
Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK, Soisson AP, Dodson MK, Tolley HD, Green CL, Zempolich KA (2006) Phosphatidylinositol 3-kinase inhibition by LY294002 radiosensitizes human cervical cancer cell lines. Clinical Cancer Res 12(1):250–256. doi:10.1158/1078-0432.CCR-05-1084
MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL (2003) Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol 79(5):351–358
Benini S, Manara MC, Cerisano V, Perdichizzi S, Strammiello R, Serra M, Picci P, Scotlandi K (2004) Contribution of MEK/MAPK and PI3-K signaling pathway to the malignant behavior of Ewing’s sarcoma cells: therapeutic prospects. Int J Cancer 108(3):358–366. doi:10.1002/ijc.11576
Badinloo M, Esmaeili-Mahani S (2014) Phosphatidylinositol 3-kinases inhibitor LY294002 potentiates the cytotoxic effects of doxorubicin, vincristine, and etoposide in a panel of cancer cell lines. Fundam Clin Pharmacol 28(4):414–422. doi:10.1111/fcp.12043
Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J (2005) Gamma-H2AX dephosphorylation by protein phosphatase 2 A facilitates DNA double-strand break repair. Mol Cell 20(5):801–809. doi:10.1016/j.molcel.2005.10.003
Wang H, Wang M, Bocker W, Iliakis G (2005) Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J Cell Physiol 202(2):492–502. doi:10.1002/jcp.20141
Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64(7):2390–2396. doi:10.1158/0008-5472.Can-03-3207
Tomimatsu N, Mukherjee B, Burma S (2009) Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. Embo Rep 10(6):629–635. doi:10.1038/embor.2009.60
Moraes MC, de Andrade AQ, Carvalho H, Guecheva T, Agnoletto MH, Henriques JA, Sarasin A, Stary A, Saffi J, Menck CF (2012) Both XPA and DNA polymerase eta are necessary for the repair of doxorubicininduced DNA lesions. Cancer Lett 314:108–118
Goldberg IH (1987) Free radical mechanisms in neocarzinostatin-induced DNA damage. Free Radic Biol Med 3(1):41–54
Li Y, Zhang P, Qiu F, Chen L, Miao C, Li J, Xiao W, Ma E (2012) Inactivation of PI3K/Akt signaling mediates proliferation inhibition and G2/M phase arrest induced by andrographolide in human glioblastoma cells. Life Sci 90(25–26):962–967. doi:10.1016/j.lfs.2012.04.044
Potter AJ, Gollahon KA, Palanca BJ, Harbert MJ, Choi YM, Moskovitz AH, Potter JD, Rabinovitch PS (2002) Flow cytometric analysis of the cell cycle phase specificity of DNA damage induced by radiation, hydrogen peroxide and doxorubicin. Carcinogenesis 23(3):389–401
Tyagi AK, Singh RP, Agarwal C, Chan DC, Agarwal R (2002) Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth Inhibition, G2-M arrest, and apoptosis. Clin Cancer Res 8(11):3512–3519
Mendelsohn J, Fan Z (1997) Epidermal growth factor receptor family and chemosensitization. J Natl Cancer Inst 89(5):341–343
Sadowski-Debbing K, Coy JF, Mier W, Hug H, Los M (2002) Caspases–their role in apoptosis and other physiological processes as revealed by knock-out studies. Arch Immunol Ther Exp 50(1):19–34
O’Gorman DM, McKenna SL, McGahon AJ, Knox KA, Cotter TG (2000) Sensitisation of HL60 human leukaemic cells to cytotoxic drug-induced apoptosis by inhibition of PI3-kinase survival signals. Leukemia 14(4):602–611
Hemmings BA, Restuccia DF (2012) PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 4(9):a011189. doi:10.1101/cshperspect.a011189
Bozulic L, Surucu B, Hynx D, Hemmings BA (2008) PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 30(2):203–213. doi:10.1016/j.molcel.2008.02.024
Li X, Lu Y, Liang K, Liu B, Fan Z (2005) Differential responses to doxorubicin-induced phosphorylation and activation of Akt in human breast cancer cells. Breast Cancer Res 7(5):R589–R597. doi:10.1186/bcr1259
Acknowledgements
The work was supported by internal funding from Latrobe Institute of Molecular Sciences and Research (LIMS) and other post graduate research funds, Latrobe University, Australia. The authors would like to thank Latrobe University for supporting the research and providing Rejitha Suraj with Postgraduate Research scholarship (LTUPS) and Full fee Research scholarship (LTUFFRS), with additional research funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Suraj, R., Radhamani, S., Meehan-Andrews, T. et al. Role of a novel benzoxazine derivative in the chemosensitization of colon cancer. Apoptosis 22, 988–1000 (2017). https://doi.org/10.1007/s10495-017-1380-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-017-1380-4