
Abstract
Ceramide-accumulation is known to be involved in the pathogenesis of chronic inflammatory lung diseases including cigarette smoke-induced emphysema (CS-emphysema) but the exact sphingolipid metabolite that initiates emphysema progression remains ambiguous. We evaluated here a novel role for the sphingolipid, lactosylceramide (LacCer), as a potential mechanism for pathogenesis of CS-emphysema. We assessed the expression of LacCer, and LacCer-dependent inflammatory, apoptosis and autophagy responses in lungs of mice exposed to CS, as well as peripheral lung tissues from COPD subjects followed by experimental analysis to verify the role of LacCer in CS-emphysema. We observed significantly elevated LacCer-accumulation in human COPD lungs with increasing severity of emphysema over non-emphysema controls. Moreover, increased expression of defective-autophagy marker, p62, in lung tissues of severe COPD subjects suggest that LacCer induced aberrant-autophagy may contribute to the pathogenesis of CS-emphysema. We verified that CS-extract treatment significantly induces LacCer-accumulation in both bronchial-epithelial cells (BEAS2B) and macrophages (Raw264.7) as a mechanism to initiate aberrant-autophagy (p62-accumulation) and apoptosis that was rescued by pharmacological inhibitor of LacCer-synthase. Further, we corroborated that CS exposure induces LacCer-accumulation in murine lungs that can be controlled by LacCer-synthase inhibitor. We propose LacCer-accumulation as a novel prognosticator of COPD-emphysema severity, and provide evidence on the therapeutic efficacy of LacCer-synthase inhibitor in CS induced COPD-emphysema.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Even though the mechanisms directing the development of chronic obstructive pulmonary disease (COPD) are not completely deciphered, toxic substances and free radicals in cigarette smoke (CS) have been shown as the leading cause for pathogenesis of emphysema in COPD subjects [1, 2]. The CS induced inflammatory-oxidative stress is clearly demonstrated as a mechanism for pathogenesis of CODP-emphysema [3–5]. Recently, we and others demonstrated the role of ceramide-accumulation as a mechanism for inflammatory-oxidative stress in cystic fibrosis (CF) [6, 7], emphysema and COPD [6, 8–11], but the exact ceramide species or sphingolipid metabolite that induces chronic lung disease or COPD-emphysema is not identified [12, 13].
Ceramide is enzymatically converted to a metabolite, lactosylceramide (LacCer) that regulates several important cellular functions, including regulation of cell proliferation, adhesion, angiogenesis and apoptosis [14, 15]. Altered expression of LacCer or LacCer-synthase (the enzyme which converts glucosylceramide to LacCer) is associated with cancer, neuroinflammatory diseases [16] and human polycystic kidney disease [17–19] but its role in pathogenesis of lung diseases such as CS induced COPD-emphysema is not investigated. Based on our recent studies on pathogenic mechanisms of lipid-raft signaling in chronic obstructive lung diseases (COPD-emphysema and CF) [6, 8], we predicted that ceramide may be leading to membrane LacCer-accumulation as a critical pathogenic step that regulates inflammation, autophagy and apoptosis responses in lung injury and emphysema. Thus, in this study, we sought to identify the specific sphingolipid involved in pathogenesis of CS induced COPD-emphysema, as it may lead to the development of effective therapeutics to alleviate chronic lung disease.
Results
LacCer accumulation correlates with severity of emphysema in COPD
To identify the role of sphingolipid, LacCer in CS induced COPD-emphysema, we procured human lung tissue and sections [non-emphysema controls (GOLD 0) and emphysema COPD (GOLD I–IV), smokers/non-smokers, Table 1)] and analyzed the statistical correlation of LacCer-accumulation with the severity of emphysema in COPD subjects. We observed a significant statistical correlation between LacCer-accumulation and increasing severity of emphysema in COPD subjects (Fig. 1a). We previously reported that CS exposure elicits a defective-autophagy response marked by perinuclear accumulation of p62 via lipid-raft ceramide-accumulation [8]. Further, we have recently reported that p62-accumulation statistically correlates with the severity of emphysema [20], similar to LacCer (Fig. 1a) suggesting that LacCer mediated aberrant-autophagy may be a novel mechanism for COPD-emphysema pathogenesis. We postulated based on this data (defective-autophagy) and known functions (apoptosis and inflammatory-oxidative stress) of LacCer that it’s increased membrane accumulation can be a potential mechanism leading to chronic lung injury and emphysema in COPD. We verified that LacCer accumulates in the membrane lipid-rafts (similar to ceramide) by co-immunostaining with lipid-raft marker, ZO-2 (Fig. 1b). This was further validated by an increased LacCer accumulation in the purified lipid-raft fractions from lung tissues of COPD subject’s (GOLD IV) with emphysema compared to non-emphysema controls (GOLD 0, Fig. 1c). We also found a significant increase in p62 expression in total protein lung extracts from emphysema subjects (COPD, GOLD IV) compared to non-emphysema controls (GOLD 0) (Fig. 1d). This was accompanied by a significant increase in p53 and CHOP expression verifying senescence and apoptosis in COPD-emphysema subjects as previously described [21, 22]. The limited number of non-smoker emphysema subjects in the study group suggests that smoking may induce membrane LacCer-accumulation, increased ZO-2 expression and perinuclear p62-accumulation (Online resource 1A) that warrants further experimental verification in human subjects (ongoing studies in larger non-smoker population), and murine and in vitro models (this study). The present data identifies a novel role of the sphingolipid, LacCer, as a potential pathogenic step for COPD-emphysema pathogenesis.

Increased LacCer accumulation correlates with severity of emphysema. a The human lung tissue sections from non-emphysema controls [(GOLD 0, n = 5, smokers (Smk) and n = 10 non-smokers (non-smk)] and COPD subjects (GOLD I–IV) with emphysema [(n = 40, smokers (Smk) and n = 5 non-smokers (non-smk)] were immunostained with LacCer. The nuclear staining (Hoechst, blue) shows the selected tissue area. The data implies a significant positive correlation (lower panel, densitometry analysis, p < 0.01) between increased LacCer with severity of emphysema in COPD subjects. b The co-immunolocalization of LacCer with lipid-raft marker, ZO-2 in the lung tissue sections (n = 60) demonstrate that LacCer-accumulation in lipid-rafts increases with severity of emphysema in COPD subjects. c The lipid-raft proteins isolated from human lung tissue from non-emphysema control (GOLD 0) and emphysema-COPD (GOLD IV) subjects were used to verify the significant increase (lower panel, densitometric analysis, p < 0.05) in raft-LacCer accumulation in severe COPD-emphysema (n = 3). The α-actin was used as a loading control. d Immunoblotting of total protein lysates from lung tissues of non-emphysema control (GOLD 0) and emphysema-COPD (GOLD IV) subjects show a significant increase in p62 (defective-autophagy marker), p53 (senescence marker) and CHOP (apoptosis marker) expression (n = 4) in the COPD-emphysema subjects compared to non-emphysema controls (p < 0.05). Scale white bar 50 μm, red bar 10 μm (Color figure online)
Cigarette smoke induces LacCer mediated aberrant-autophagy and apoptosis
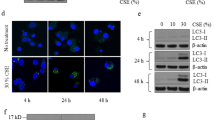
LacCer is an important signaling intermediate for the induction of pro-inflammatory mediators and astrogliosis [16, 23]. The activation of NADPH oxidase and NFκB signaling cascades are believed to be involved [15, 23] in LacCer mediated inflammatory signaling. We have previously shown that CS induces inflammation, apoptosis and aberrant-autophagy in murine lungs and airways cells [8, 20, 21]. To verify whether CS induces LacCer-accumulation, and if LacCer is a critical step for inducing inflammatory-apoptotic responses and defective-autophagy, we used human bronchial epithelial (BEAS2B) and macrophage (Raw264.7) cells treated with CS-extract (CSE) and murine lung injury and emphysema models [C57BL/6 mice exposed to acute-, sub-chronic- or chronic-CS and Pseudomonas aeruginosa Lipopolysaccharide (Pa)-LPS]. We further substantiated the role of LacCer in COPD-emphysema pathogenesis using the pharmacological inhibitors of LacCer-synthase (d-PDMP or PBPP) in these models followed by functional analysis of inflammatory, apoptotic and aberrant-autophagy responses. We first confirmed that CSE treatment significantly increases LacCer-accumulation in both epithelial (BEAS2B) cells and macrophages (Raw264.7) (Fig. 2a). Next, we observed that inhibiting LacCer-accumulation by d-PDMP and PBPP was effective in significantly reducing the number of CSE-induced p62-positive cells (Fig. 2b). Since CSE induces LacCer-accumulation and aberrant-autophagy, we confirmed whether LacCer treatment could increase the number of p62-positive cells over other structurally related molecules (controls) (Fig. 2c). We also evaluated the specificity of pharmacological inhibitor of LacCer-synthase (d-PDMP) in controlling LacCer synthesis and accumulation (Fig. 2d). Accumulation of p62 along with the autophagosome marker-LC3β and ubiquitinated protein aggregates (aggresome) is considered as a functional marker of defective-autophagy [24]. Thus co-localization of LC3-GFP and Ubiquitin-RFP in Raw264.7 cells was used as a functional reporter to access if CSE induces aberrant-autophagy via LacCer. As anticipated, CSE induces perinuclear co-localization of LC3-GFP and Ub-RFP (aggresome) while d-PDMP inhibits LC3-Ub accumulation (Fig. 2e). Since LacCer-synthase inhibitors decreased the number of CSE-induced p62-positive cells (Fig. 2b) and LC3-Ub accumulation (Fig. 2e) we confirm that LacCer plays a crucial role in regulating CSE-induced autophagy response. A recent study demonstrates the crucial role of LacCer in NSMase induced apoptosis [25]. We verified here that CSE induces apoptosis via LacCer as seen by a significant increase in sub-G1 fraction (M1) in the presence of CSE that can be rescued to basal levels with LacCer-synthase inhibitors (Fig. 2f).

Cigarette smoke extract (CSE) induces LacCer mediated aberrant-autophagy and apoptosis. a The BEAS2B- and Raw264.7-cells were treated with DMSO (control) or cigarette smoke extract (CSE, 200 μg/ml, 24 h) and the increase in percentage of LacCer-positive cells was quantified by flow cytometry. CSE treatment shows a very significant (p < 0.001) increase in LacCer-accumulation (34-fold, BEAS2B and 99.7-fold, Raw264.7 cells). b The Raw264.7 cells were treated with DMSO (control), CSE (200 μg/ml) and/or LacCer-synthase inhibitors (d-PDMP & PBPP, 50 μM) for 24 h and the percentage of p62-positive cells were analyzed by flow cytometry. The data indicates that d-PDMP and PBPP efficiently control the number of CSE induced aberrant-autophagic cells. c The Raw264.7 cells were treated with LacCer, dihydro-lactosylceramide (dLac) or di-galactosylceramide (di-Gal) (2 μg/ml each) for 12 h and the percentage of p62-postive cells in treatment group as compared to no-treatment control was analyzed by flow cytometry. The data indicate the specificity (p < 0.01) of LacCer in inducing aberrant-autophagy as compared to other structurally related molecules (dLac, diGal). d Flow cytometry of Raw264.7 cells shows the specificity of pharmacological inhibitor of LacCer-synthase, d-PDMP (50 μM, 12 h) in controlling the LacCer expression (p < 0.01). e Immunofluorescence microscopy of Raw264.7 cells transiently transfected with LC3-GFP and Ub-RFP plasmids for 12 h followed by DMSO (control) or CSE (200 μg/ml) treatment for 24 h shows increased number of LC3-Ub-positive bodies (white arrows) after CSE treatment. Moreover, treatment with LacCer synthase inhibitor, d-PDMP (50 μM), significantly (n = 3, p < 0.01) controls the number of CSE induced LC3-Ub-positive bodies. At least three independent experiments were performed, and the representative data is shown. f The apoptotic changes in murine peritoneal macrophages, treated in vitro with CSE (200 μg/ml) and/or LacCer-synthase inhibitors, d-PDMP/PBPP (50 μM, 12 h) was analyzed by propidium iodide (PI) staining. Data shows a significant reduction (p < 0.001) in percentage of CSE induced apoptotic cells (M1-sub-G1 fraction) upon treatment with LacCer-synthase inhibitors (n = 3)
LacCer mediates CS or Pa-LPS induced emphysema and lung inflammation
Having identified lipid-raft LacCer-accumulation as a potential mechanism for inflammatory-oxidative stress, apoptosis and aberrant-autophagy induction, we authenticated these findings in murine models of CS and Pseudomonas aeruginosa-LPS (Pa-LPS) induced lung injury. Recent studies show that both acute-CS and LPS induce lung injury and emphysema-like phenotype in mice [3]. Subsequently, we found that acute-CS/Pa-LPS exposure of C57BL/6 mice induced LacCer-accumulation in murine lungs (Fig. 3a, e). The functional role of LacCer in modulating inflammation, apoptosis and autophagy responses during lung injury was verified by using pharmacological inhibitors of LacCer-synthase, d-PDMP or PBPP. We found that treatment with d-PDMP and PBPP successfully controlled acute-CS; sub-chronic CS/Pa-LPS induced lung inflammation, apoptosis and aberrant-autophagy (Figs. 3, 4; Online resource 1B, C).

LacCer mediates cigarette smoke (CS) or Pa-LPS induced lung inflammation and apoptosis. a Immunostaining shows increased LacCer-accumulation (red) in longitudinal lung tissue sections from acute-CS (A-CS, 1 week) exposed C57BL/6 mice (n = 3) compared to air controls. b–d Treatment of acute-CS-exposed mice with pharmacological inhibitors of LacCer-synthase [d-PDMP and PBPP, 50 μg/mouse, intra-tracheal (i.t), 24 h] controls acute-CS induced lung inflammation [(b H&E, c BALF-interleukin-6 (IL-6)] and apoptosis (d lung-caspase-3/7 activity). e Immunostaining shows increased LacCer-accumulation (red) in longitudinal lung tissue sections from Pa-LPS (20 μg/mouse, 36 h) treated C57BL/6 mice (n = 3) compared to PBS controls. Treatment with LacCer-synthase inhibitors (50 μg/mouse) significantly control Pa-LPS induced inflammation (f H&E and g BALF-IL-6 levels) and apoptosis (h lung-caspase-3/7 activity) (n = 3, p < 0.05). Data represents at least triplicate independent experiments. Scale white/black bar 50 μm (Color figure online)

LacCer-synthase inhibition ameliorates sub-chronic-CS induced lung injury and aberrant-autophagy. a The longitudinal lung tissue sections of air- or sub chronic-CS (SC-CS)-exposed (4 weeks) and/or d-PDMP [50 μg/mouse, intra-tracheal (i.t.) for the last 24 h] treated C57BL/6 mice were either stained with H&E to detect the inflammatory state, or immunostained with NFκB, F4/80, NIMP R-14 and p62 polyclonal antibodies. Subchronic-CS-exposure significantly induces the levels of the inflammatory (NFκB, F4/80, NIMP R-14) and defective-autophagy marker (p62) that are controlled by treatment with d-PDMP (right panels, densitometric analysis, p < 0.05). n = 3, Scale white/black bar 50 μm. The nuclear staining (Hoechst) in the bottom panel of each staining shows the selected tissue area. b The subchronic CS-exposed mice show a significant increase in bronchoalveolar fluid (BALF) IL-6 levels (p = 0.029) that are controlled by LacCer-synthase inhibitor, d-PDMP (p < 0.01, n = 3). c The flow cytometry analysis of BALF-cells isolated from subchronic-CS-exposed and/or d-PDMP treated mice show that LacCer-synthase inhibition significantly (p < 0.01) controls subchronic-CS induced macrophage (Mac-3-positive cells) infiltration
To further validate if CS induced LacCer-accumulation leads to the development of emphysema we exposed C57BL/6 mice to chronic-CS and treated with d-PDMP as per the experimental timeline shown in Fig. 5a. We found that chronic-CS exposure significantly induces LacCer-accumulation in the lungs that correlate with severity of emphysema (Fig. 5b, e). Moreover, accumulation of ubiquitinated proteins and increased expression of defective-autophagy marker (p62) (Fig. 5d) was observed with Chronic-CS exposure. We verified the role of LacCer in emphysema pathogenesis using the pharmacological inhibitor of LacCer-synthase, d-PDMP that not only ameliorates chronic-CS induced inflammation but can also control CS induced alveolar airspace enlargement (Fig. 5e, f). We also found that level of pro-inflammatory cytokine, IL-6 in bronchoalveolar lavage fluid (BALF) of chronic-CS exposed mice was significantly decreased by d-PDMP treatment (Fig. 5g). Overall, we observed that d-PDMP treatment can control CS-induced chronic inflammation, alveolar airspace enlargement, macrophage infiltration (Fig. 5) and defective-autophagy (Fig. 6).

LacCer-synthase inhibitor ameliorates chronic-CS induced lung inflammation and emphysema. a The experimental timeline for treatments with LacCer-synthase inhibitor (d-PDMP; 10 μg/mouse, n = 4–5) in chronic-CS induced emphysema murine model. The longitudinal lung tissue sections from mice exposed to room air- or chronic-CS shows a considerable increase in LacCer-accumulation upon chronic-CS-exposure (b) that is significantly downregulated by d-PDMP treatment (c; p < 0.05). The nuclear staining (Hoechst, blue) in the bottom panel shows the selected tissue area. d The total lung protein lysates from chronic-CS exposed mice show an increase in p62 expression compared to air exposed mice, indicating a defective-autophagy response that is verified by an increase in ubiquitin-accumulation. β-actin was used as the loading control. e The H&E staining of these lung tissue sections show a decrease in CS induced inflammation on treatment with d-PDMP. f The morphometric analysis of lung tissue sections stained with H&E (e) demonstrate a significant increase in alveolar diameter upon chronic-CS exposure (1.55-fold, p = 0.000017). This chronic-CS induced alveolar space enlargement is significantly inhibited (1.55 to 1.23-fold, p = 0.0016) by LacCer-synthase inhibitor (d-PDMP). g The chronic-CS exposed mice show a significant increase in BALF IL-6 levels (p = 0.01) that can be rescued to basal levels by treatment with d-PDMP (n = 3, p = 0.009). h The flow cytometric analysis of bronchoalveolar fluid (BALF)-cells isolated from chronic-CS exposed and/or d-PDMP treated mice shows that d-PDMP treatment significantly controls (p < 0.01) chronic-CS induced activated-macrophage (F4/80-NFκB-positive cells) infiltration. Data represents at least triplicate independent experiments. Scale white/black bar 50 μm

Chronic-CS induces inflammation and aberrant-autophagy via LacCer. a The longitudinal lung tissue sections of air- or chronic-CS (Ch-CS)-exposed and/or d-PDMP treated mice were either immunostained with p62, NFκB, F4/80, NIMP R-14 polyclonal antibodies. The nuclear staining (Hoechst) in the bottom panel shows the selected tissue area. The data indicate that treatment with LacCer synthase inhibitor (d-PDMP) significantly (right panels p < 0.05, densitometric analysis) ameliorates chronic-CS induced inflammation and chronic lung injury
We anticipate based on these and our previous study [6] that CS-exposure modulates lipid-raft clustering by inducing LacCer accumulation via CFTR, as Cftr −/− mice intrinsically show increased LacCer accumulation (Fig. 7a). This clustering of lipid-rafts is known to augment apoptotic and NFκB mediated pro-inflammatory signaling through TNFR, IL-1R, TLR and/or FAS receptors [8, 26–28]. LacCer is known to be involved in inflammatory-oxidative signaling [15] and LPS/IFNγ induced iNOS generation [23]. Our data also suggest that CS induces aberrant proteostasis [21] that leads to Ub-p62-dependent perinuclear aggregation of misfolded proteins (aggresomes). Intriguingly, CFTR is essential to maintain a robust autophagy response to clear-off p62-positive aggresomes that accumulate in CF lung disease [24]. We predict that defective-autophagy [20], inflammation and apoptosis, all contribute to CS-LacCer mediated emphysema. Additionally, increase in membrane-LacCer-accumulation recruit’s caveolin-1 to lipid-raft microdomains [24] that in turn may enhance the susceptibility to LacCer induced emphysema by p53-dependent cellular senescence mechanisms [22, 29] (Fig. 7b).

Absence of CFTR triggers LacCer-accumulation. a The longitudinal lung sections from Cftr −/− mice lungs show a significant increase (lower panel, densitometric analysis, p < 0.001) in LacCer-accumulation as compared to Cftr +/+ mice lungs. The nuclear staining (Hoechst) in the middle panel shows the selected tissue area. The data indicate that absence of CFTR induces LacCer-accumulation. Scale white bar 50 μm. b Schematic elucidating the proposed mechanisms of LacCer mediated chronic lung injury and emphysema in COPD. Cigarette smoke exposure decreases lipid-raft CFTR that leads to accumulation of a novel sphingolipid, LacCer. This promotes clustering of lipid-rafts that augments apoptotic and NFκB mediated pro-inflammatory signaling via TNFR, IL-1R, TLR and/or FAS receptors. Moreover, CS induces aberrant proteostasis that leads to Ub-p62-dependent, misfolded protein accumulation as perinuclear aggregates (aggresomes). The accumulation of p62+ aggresomes indicate defective-autophagy that contributes to CS induced COPD-emphysema pathogenesis. Additionally, increase in membrane-LacCer- accumulation may recruit caveolin-1 to lipid-raft microdomains that in turn may enhance the susceptibility to LacCer induced emphysema by p53-dependent cellular senescence mechanisms
To summarize, we demonstrate here that LacCer-accumulation is a common pathogenic intermediate that regulates apoptotic-inflammatory responses and aberrant-autophagy, as a mechanism for COPD-emphysema pathogenesis. Moreover, we demonstrate the therapeutic-efficacy of pharmacological LacCer-synthase inhibitors in controlling chronic-CS induced emphysema.
Discussion
The pathogenesis of chronic inflammatory lung diseases including CS induced emphysema is proposed to involve lipid-raft ceramide-accumulation [6–8] but the exact sphingolipid metabolite that initiates emphysema progression remains to be identified. In this study we identified and verified a novel role of the sphingolipid, LacCer in CS-induced COPD-emphysema. We found that CS induced LacCer-accumulation is a common pathogenic mechanism that induces apoptotic-inflammatory responses and aberrant-autophagy leading to severe emphysema in COPD.
We and others have recently elucidated the critical role of lipid-raft signaling platforms in mediating chronic inflammatory-apoptotic responses and lung injury in both CF and COPD [6–8]. LacCer is a lipid-raft associated glycosphingolipid (GSL) [30] that is known to be involved in inflammatory-oxidative signaling [15] and LPS/IFNγ induced iNOS generation [23]. We anticipated based on these and our recent studies [6, 8] that CS-exposure modulates lipid-raft clustering by inducing LacCer-accumulation via CFTR (Fig. 7a). This clustering of lipid-rafts may augment apoptotic and NFκB mediated pro-inflammatory signaling through TNFR, IL-1R, TLR and/or FAS receptors [8, 26–28]. As anticipated, our data demonstrates a positive statistical correlation of LacCer-accumulation with increasing severity of emphysema in COPD lungs (Fig. 1a). Moreover, we also verified the expression of known apoptotic and senescence related proteins, CHOP (GADD153) and p53 [21–23, 31] with emphysema progression (Fig. 1d) suggesting that LacCer-accumulation may induce inflammatory-apoptotic signaling leading to chronic lung disease and/or severe emphysema. Next, we experimentally verified that CSE/CS can significantly induce LacCer-accumulation in BEAS2B/Raw264.7 cells and murine lungs (acute/chronic-CS/Pa-LPS) (Figs. 2, 3, 4, 5, 6). Moreover, we confirmed the functional role of CS-induced LacCer-accumulation in inducing inflammatory-apoptotic signaling by using pharmacological inhibitors of LacCer-synthase (d-PDMP/PBPP) (Figs. 2, 3, 4, 5, 6). These LacCer-synthase inhibitors have been previously used to validate the role of LacCer in cell migration and proliferation of human aortic smooth muscle cells [14], TNFα-induced proliferation of rat primary astrocytes [16] and the lipopolysaccharide/interferon-γ-mediated inducible nitric oxide synthase gene expression [23]. As discussed above, we treated the CSE/CS-exposed cells and mice with d-PDMP and PBPP and found their efficacy in controlling CS-induced inflammation and apoptosis. Our human- and murine-COPD-emphysema data suggests the role of significantly elevated LacCer-accumulation in inflammatory-apoptotic signaling similar to previous reports for other disease states such as cancer [17, 32], neuroinflammatory conditions [23], atherosclerogenesis and atherosclerosis [14]. LacCer is also known to mediate TNFα-induced superoxide/ROS production from neutrophils and endothelial cells [15, 33]. Moreover, treatment with the specific inhibitors of LacCer-synthase, d-PDMP or PBPP abates LacCer-mediated inflammation, cancer cell migration and oxidative damage [17, 25, 34]. Another recent report shows that reducing GSL biosynthesis by treatment with anti-sense oligonucleotides against GSL synthesis enzymes, including the inhibition of LacCer-synthase, in airway cells partially ameliorates disease outcomes in mouse model of allergic asthma [35]. Our current data not only suggests a novel role of LacCer in the pathogenesis of CS-induced lung injury and emphysema but also demonstrate the therapeutic potential of LacCer synthase inhibitors in controlling CS induced COPD-emphysema.
We recently reported that the cytoplasmic accumulation of misfolded/polyubiquitinated proteins in aggresome bodies in response to CS-exposure induces inflammatory-apoptotic signaling [20, 21]. We predict that CS induced aggresome formation may be a result of autophagy-dysfunction since autophagy is a cytoprotective mechanism that participates in the clearance of misfolded/polyubiquitinated proteins by targeting them for degradation [24]. A recent study by Choi et al. reported elevated autophagy in the lungs of COPD patients and CS-exposed murine lungs [36]. They proposed a crucial role of autophagic protein, LC3β in mediating CS-induced apoptosis and emphysema [36]. Our current and previous studies suggests that autophagy dysfunction leads to COPD-emphysema pathogenesis and clearly support that CS induced aberrant-autophagy [21] leads to Ub-p62-dependent perinuclear aggregation of misfolded proteins (aggresomes) that mediates chronic inflammation, apoptosis and emphysema. Since autophagy is central to clearance of aggresomes, we propose that defective-autophagy is a critical mechanism of CS-induced lung injury and emphysema. In accord with this, we observed an accumulation of aberrant-autophagy markers, p62/Ub [21] in COPD-emphysema subjects and CS-exposed murine lungs ([20]; Figs. 4, 6; Online resource 1B). We verified that CSE-induces perinuclear co-localization of LC3-GFP and Ub-RFP (indicator of aberrant-autophagy) that can controlled by d-PDMP treatment (Fig. 2e) supporting the critical role of LacCer in regulating CSE induced aberrant-autophagy. Intriguingly, CFTR is also essential to maintain a robust autophagy response to clear-off p62-positive aggresomes that accumulate in CF lung disease [24]. We and others have shown that CS down regulates CFTR expression and function [6, 37] and this could be one potential mechanism for CS-induced aberrant-autophagy (Fig. 7b). Although, it is also possible that CS directly influences autophagy [20] in addition to CFTR dependent mechanism. Our recent study demonstrates a significant increase in constitutive and CS-induced p62- and LC3β-expression in the CFTR-deficient cells and mice [8], supporting the role of CFTR in controlling autophagy responses in CS-induced lung injury and emphysema. In the present study, we found that Cftr −/−mice lungs have higher LacCer-accumulation compared to Cftr +/+ mice (Fig. 7a). We predict that CS directs aberrant-autophagy via its effect on membrane-CFTR expression and function [24, 38] that controls lipid-raft LacCer-accumulation. Our in vitro experiments in Raw264.7 cells validate that CS induces LacCer-accumulation that leads to p62-accumulation (Fig. 2a–c). This implies that CS exposure promotes defective-autophagy response through LacCer-accumulation. Based on our in vitro and in vivo functional rescue experiments with specific LacCer-synthase inhibitors (d-PDMP/PBPP) we propose that controlling LacCer-accumulation may mitigate the CS-induced aberrant-autophagy [20, 21] and inflammatory-apoptotic signaling that contribute to CS-LacCer mediated COPD-emphysema pathogenesis.
Overall, LacCer-accumulation in human and murine emphysema and its association with severity of lung disease demonstrates its potential utility as a prognosticator of emphysema state and aforementioned therapeutic strategy for COPD-emphysema subjects.
Materials and methods
Human subjects and murine experiments
Frozen lung tissue samples and longitudinal sections were obtained from the NHLBI Lung Tissue Research Consortium (LTRC, NIH). The clinical severity, sample size and classification of COPD lung disease subjects was graded on the basis of stages defined by Global Initiative for Chronic Obstructive Lung Disease (GOLD) [Control (GOLD 0) and COPD (GOLD I–IV), n = 10–15 for each stage, see Table 1 [39, 40]. None of the control- or COPD-subjects had any other underlying condition other than emphysema for COPD subjects (Gold I–IV). Moreover, one patient in each group (Gold I–IV) had first-degree blood relatives with chronic bronchitis. The study protocol was approved by the Institutional Review Board (IRB), Johns Hopkins University as exempt, and subject’s lung function data and other clinical parameters were obtained from each of the LTRC contributing centers without disclosing the subject’s name and information. The lung samples were analyzed by immunostaining or immunoblotting and statistical correlation of the data was calculated (described below). The results of the human data were experimentally verified using CS and Pa-LPS induced emphysema and lung injury murine models. All animal experiments were carried out in accordance with the Johns Hopkins University (JHU) Animal Care and Use Committee (ACUC) approved protocols. We used age, weight and sex matched (8–10 weeks old) C57BL/6 mice (NCI Animal Production Program and in house breeding), n = 3–4 for all experiments. All mice were housed in controlled environment and pathogen-free conditions. Mice were exposed to room-air (control) or acute-(1 week), sub-chronic-(4 weeks) or chronic-(24 weeks) CS using the TE-2 cigarette smoking machine [6, 8, 21] (Teague Enterprises, Davis, CA). The CS was generated by burning research-grade cigarettes (3RF4; 0.73 mg nicotine per cigarette) purchased from the Tobacco Research Institute (University of Kentucky, Lexington, KY) for 3 h/day direct smoke followed by 2 h in the same chamber resulting in an average total particulate matter of 150 mg/m3. The air- and CS-exposed mice were treated intra-tracheally with 50 µg/mouse d-PDMP/PBPP (details in ‘in vitro and ex vivo experiments’; Matreya LLC, Pleasant Gap, PA) for acute and sub-chronic CS/air-exposure experiments. In a separate experiment, mice were treated with Pa-LPS (Sigma, St. Louis, MO, intra-tracheal; 20 µg/mouse) for 36 h [6] and/or d-PDMP/PBPP (50 µg/mouse) for the last 24 h. We assessed the dose dependent efficacy of d-PDMP in Pa-LPS murine model and identified 10 µg/mouse d-PDMP as a potent minimal dose that can control both inflammation and aberrant-autophagy. The selected dose was given to the mice at the indicated time points (Fig. 5a) to test its efficacy in controlling chronic-CS induced emphysema. To evaluate the effect of CFTR-deficiency on LacCer-accumulation, we used the Cftr −/− murine model as recently described [6, 8].
In vitro and ex vivo experiments
The BEAS2B (human bronchial epithelial cells) and Raw264.7 (human macrophages) cells were cultured at 37 °C/5 % CO2 in DMEM F-12 and RPMI media respectively, supplemented with 10 % fetal bovine serum (FBS) and 1 % penicillin, streptomycin and amphotericin B (PSA), all from Invitrogen. The cells were treated with CSE (200 µg/ml, Murty Pharmaceuticals), LacCer, dihydrolactosylceramide (dLac), di-galactosyldiglyceride (diGal) (2 µg/ml, Matreya, LLC), LacCer-synthase inhibitors, d-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (d-PDMP)/d-threo-1-phenyl-2-benzyloxycarbonylamino 3pyrrolidino-1-propanol (PBPP) (50 μM, Matreya, LLC) for 24 h. After these treatments the cells were fixed using 1 % paraformaldehyde (USB, Cleveland, OH) and washed in FACS buffer (1X, 2 % FBS in PBS). The non-specific antibody binding was blocked by incubating the cells with either donkey or goat serum (1:10; Sigma) for 30 min. The cells were stained with LacCer (rabbit polyclonal, Abbiotec, San Diego CA) or p62 (mouse monoclonal, BD Biosceinces, Franklin Lakes, NJ) primary antibodies followed by anti-rabbit FITC or anti-mouse phycoerythrin (PE) secondary antibodies (Invitrogen) for 1 h, respectively. The FIX& PERM ™ cell permeabilization kit (Invitrogen) was used for p62 and NFκB intracellular staining following the manufacturer’s protocol. Similarly, BALF-cells isolated from sub-chronic- or chronic-CS-exposed and/or d-PDMP treated mice were fixed and stained with F4/80 (rat monoclonal, scbt), Mac-3 (rat monoclonal, Abcam) and/or NFκB-FITC (scbt) primary antibodies followed by anti-rat R-PE secondary antibody (Invitrogen). Finally, cells were washed in FACS buffer (1X), resuspended in 0.1 % paraformaldehyde and data was acquired and analyzed using the BD FACS Caliber instrument and BD Cell Quest Pro software. Appropriate secondary antibody controls (anti-rat-PE, anti-rabbit-FITC, anti-mouse-PE) were used in all the flow cytometry experiments. For quantifying effect of LacCer synthase inhibitors on apoptosis, thioglycolate-elicited murine peritoneal macrophages were treated with CSE (200 µg/ml) and/or d-PDMP/PBPP (50 μM) for 24 h and fixed in ice cold 10 % ethanol followed by propidium iodide (PI) staining [41]. The data was acquired using the BD FACS Caliber instrument and the number of apoptotic cells (M1-sub-G1 fraction) were quantified using the BD Cell Quest Pro software.
Immunostaining, microscopy, lipid-raft isolation, immunoblotting, ELISAs and apoptosis-autophagy assays
Detailed methods in online resource 3.
Statistical analysis
Data is represented as the mean ± SD (flow cytometry, immunostaining, autophagy reporter assay) or SEM (lung morphometry, ELISA, caspase 3/7 assay, western blotting) of at least three experiments. The Student’s t test and 2-way ANOVA were used to determine the statistical significance. The murine and human microscopy data were analyzed by densitometry (Matlab R2009b, Mathworks Co.) followed by for Spearman’s correlation coefficient analysis to calculate the significance among the indicated groups. While the total number of apoptotic, inflammatory and autophagic cells were quantified by TUNEL, immunostaining and autophagy reporter assays followed by Spearman’s correlation coefficient analysis to calculate the significance among the indicated groups.
References
Barnes PJ (2003) New concepts in chronic obstructive pulmonary disease. Annu Rev Med 54:113–129
MacNee W (2005) Oxidants and COPD. Curr Drug Targets Inflamm Allergy 4(6):627–641
van Houwelingen AH, Weathington NM, Verweij V, Blalock JE, Nijkamp FP, Folkerts G (2008) Induction of lung emphysema is prevented by l-arginine-threonine-arginine. FASEB J 22(9):3403–3408
Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, Shastry S, Rowe SM, Shim YM, Hussell T, Blalock JE (2010) A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science 330(6000):90–94
Barnes PJ, Shapiro SD, Pauwels RA (2003) Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 22(4):672–688
Bodas M, Min T, Mazur S, Vij N (2011) Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J Immunol 186(1):602–613
Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G, Gulbins E (2008) Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 14(4):382–391
Bodas M, Min T, Vij N (2011) Critical role of CFTR dependent lipid-rafts in cigarette smoke induced lung epithelial injury. Am J Physiol Lung Cell Mol Physiol 300(6):L811–L820
Elias JA, Lee CG (2005) Lipid let loose in pulmonary emphysema. Nat Med 11(5):471–472
Uhlig S, Gulbins E (2008) Sphingolipids in the lungs. Am J Respir Crit Care Med 178(11):1100–1114
Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM (2005) Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 11(5):491–498
Brodlie M, McKean MC, Johnson GE, Gray J, Fisher AJ, Corris PA, Lordan JL, Ward C (2010) Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am J Respir Crit Care Med 182(3):369–375
Guilbault C, De Sanctis JB, Wojewodka G, Saeed Z, Lachance C, Skinner TA, Vilela RM, Kubow S, Lands LC, Hajduch M, Matouk E, Radzioch D (2008) Fenretinide corrects newly found ceramide deficiency in cystic fibrosis. Am J Respir Cell Mol Biol 38(1):47–56
Mu H, Wang X, Wang H, Lin P, Yao Q, Chen C (2009) Lactosylceramide promotes cell migration and proliferation through activation of ERK1/2 in human aortic smooth muscle cells. Am J Physiol Heart Circ Physiol 297(1):H400–H408
Arai T, Bhunia AK, Chatterjee S, Bulkley GB (1998) Lactosylceramide stimulates human neutrophils to upregulate Mac-1, adhere to endothelium, and generate reactive oxygen metabolites in vitro. Circ Res 82(5):540–547
Pannu R, Singh AK, Singh I (2005) A novel role of lactosylceramide in the regulation of tumor necrosis factor alpha-mediated proliferation of rat primary astrocytes. Implications for astrogliosis following neurotrauma. J Biol Chem 280(14):13742–13751
Chatterjee S, Pandey A (2008) The Yin and Yang of lactosylceramide metabolism: implications in cell function. Biochim Biophys Acta 1780(3):370–382
Chatterjee S, Shi WY, Wilson P, Mazumdar A (1996) Role of lactosylceramide and MAP kinase in the proliferation of proximal tubular cells in human polycystic kidney disease. J Lipid Res 37(6):1334–1344
Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, Belenky A, Bukanov NO, Dackowski WR, Husson H, Russo RJ, Shayman JA, Ledbetter SR, Leonard JP, Ibraghimov-Beskrovnaya O (2010) Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med 16(7):788–792
Tran I, Ji C, Ni I, Min T, Tang D, Vij N (2014) Role of cigarette smoke-induced aggresome-formation in COPD-emphysema pathogenesis. Am J Respir Cell Mol Biol. doi:10.1165/rcmb.2014-0107OC
Min T, Bodas M, Mazur S, Vij N (2011) Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J Mol Med (Berl) 89(6):577–593
Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW (2006) Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol 35(6):681–688
Pannu R, Won JS, Khan M, Singh AK, Singh I (2004) A novel role of lactosylceramide in the regulation of lipopolysaccharide/interferon-gamma-mediated inducible nitric oxide synthase gene expression: implications for neuroinflammatory diseases. J Neurosci 24(26):5942–5954
Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, D’Apolito M, Guido S, Masliah E, Spencer B, Quaratino S, Raia V, Ballabio A, Maiuri L (2010) Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 12(9):863–875
Martin SF, Williams N, Chatterjee S (2006) Lactosylceramide is required in apoptosis induced by N-Smase. Glycoconj J 23(3–4):147–157
Zhang Y, Li X, Becker KA, Gulbins E (2009) Ceramide-enriched membrane domains—structure and function. Biochim Biophys Acta 1788(1):178–183
Muppidi JR, Tschopp J, Siegel RM (2004) Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21(4):461–465
Grassme H, Cremesti A, Kolesnick R, Gulbins E (2003) Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 22(35):5457–5470
Volonte D, Galbiati F (2009) Caveolin-1, cellular senescence and pulmonary emphysema. Aging (Albany NY) 1(9):831–835
McDonald G, Deepak S, Miguel L, Hall CJ, Isenberg DA, Magee AI, Butters T, Jury EC (2014) Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. The J Clin Invest 124(2):712–724. doi:10.1172/JCI69571
Vij N, Amoaka MO, Mazur S, Zeitlin P (2008) CHOP transcription factor mediates IL-8 signaling in cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol 38(2):176–184
Aouali N, El Btaouri H, Dumontet C, Eddabra L, Malagarie-Cazenave S, Madoulet C, Morjani H (2011) Accumulation of lactosylceramide and overexpression of a PSC833-resistant P-glycoprotein in multidrug-resistant human sarcoma cells. Oncol Rep 25(4):1161–1167
Bismuth J, Chai H, Lin PH, Yao Q, Chen C (2009) Lactosylceramide causes endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Med Sci Monit 15(9):270–274
Won JS, Singh AK, Singh I (2007) Lactosylceramide: a lipid second messenger in neuroinflammatory disease. J Neurochem 103(Suppl 1):180–191
Karman J, Tedstone JL, Gumlaw NK, Zhu Y, Yew N, Siegel C, Guo S, Siwkowski A, Ruzek M, Jiang C, Cheng SH (2010) Reducing glycosphingolipid biosynthesis in airway cells partially ameliorates disease manifestations in a mouse model of asthma. Int Immunol 22(7):593–603
Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM (2010) Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA 107(44):18880–18885
Cantin AM, Hanrahan JW, Bilodeau G, Ellis L, Dupuis A, Liao J, Zielenski J, Durie P (2006) Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med 173(10):1139–1144
Vij N, Downey GP (2013) The yin and yang of cystic fibrosis transmembrane conductance regulator function: implications for chronic lung disease. Am J Respir Crit Care Med 187(2):120–122
Keller CA (2003) Pathophysiology and classification of emphysema. Chest Surg Clin N Am 13(4):589–613
Fromer L, Cooper CB (2008) A review of the GOLD guidelines for the diagnosis and treatment of patients with COPD. Int J Clin Pract 62(8):1219–1236. doi:10.1111/j.1742-1241.2008.01807.x
Riccardi C, Nicoletti I (2006) Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 1(3):1458–1461
Acknowledgments
NV was supported by Flight Attendants Medical Research Institute (FAMRI) and National Institute of Health (CTSA UL RR 025005, RHL096931 and U54CA141868) Grants. We would like to thank Lung Tissue Research Consortium LTRC (NHLBI, NIH) for human lung tissue samples.
Conflict of interest
The authors declare that they have no conflict of interests.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bodas, M., Min, T. & Vij, N. Lactosylceramide-accumulation in lipid-rafts mediate aberrant-autophagy, inflammation and apoptosis in cigarette smoke induced emphysema. Apoptosis 20, 725–739 (2015). https://doi.org/10.1007/s10495-015-1098-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-015-1098-0