
Abstract
Ticks serve as vectors and reservoirs of various Borrelia species, potentially causing diseases in humans and animals. Mazandaran, a fertile green land in northern Iran, provides ample grazing grounds for livestock and harbors at least 26 hard tick species. This study investigated Borrelia infection in hard ticks from forest areas in this region and compared their genetic identity with the species data in the GenBank database. A total of 2,049 ticks were collected manually from mammalian hosts or using dragging and flagging methods. These ticks were then grouped into 190 pools and 41 individuals based on host, species, developmental stage, and gender. A real-time PCR (qPCR) detected Borrelia DNA in 26 pools from female, male, and nymph of Rhipicephalus annulatus (n = 17) and Ixodes ricinus (n = 9) ticks and one individual female Haemaphysalis punctata tick. The generated partial flaB and glpQ sequences from qPCR-positive Rh. annulatus ticks exhibited the highest identities of 98.1-100% and 98.2% with Borrelia theileri and closely related undefined isolates. Additionally, in phylogenetic analysis, these sequences clustered within well-supported clades with B. theileri and the closely related undefined isolates from various geographic regions, confirming the presence of B. theileri in the north of Iran. Divergence in B. theileri flaB and glpQ sequences across various geographical areas suggests potential subspeciation driven by adaptations to different tick species. This divergence in our flaB sequences implies the possible introduction of B. theileri-infected ticks from different geographical origins into Iran.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Borrelia comprises arthropod-borne spirochetes that complete their life cycle in vertebrate hosts (Qiu et al. 2021). Phylogenetically, they are divided into three groups: Lyme Group (LG), Relapsing Fever Group (RFG), and Echidna-Reptile Group (REPG). Among these, only the LG and certain members of the RFG borreliae are known to cause infections in humans. The REPG borreliae represent a distinct monophyletic group primarily infecting amphibians and reptiles (Trevisan et al. 2021). Apart from the louse-adapted Borrelia recurrentis, most members of RFG pathogenic to humans are transmitted by argasid soft ticks (Cutler et al. 2017; Talagrand-Reboul et al. 2018). Additionally, Ixodes and other hard ticks maintain Borrelia species such as Borrelia miyamotoi, B. lonestari, B. theileri, and some undescribed species genetically grouped with RFG (Cutler et al. 2017; Furuno et al. 2017; Naddaf et al. 2020; Talagrand-Reboul et al. 2018).
In Iran, molecular analyses have identified the presence of at least two soft tick relapsing fever (STRF) borreliae: the well-established B. persica (Marti Ras et al. 1996; Naddaf et al. 2015; Shirani et al. 2016) and a complex group infecting Ornithodoros ticks, rodents, and humans with the highest resemblance to East African Borrelia duttonii and B. recurrentis (Ghasemi et al. 2021; Houmansadr et al. 2020; Naddaf et al. 2012, 2015, 2017). Recently, in north Iran, LG borreliae, Borrelia afzelii, B. garinii, B. valaisiana, B. bavariensis, and RFG B. miyamotoi were identified in Ix. ricinus ticks (Naddaf et al. 2020).
In the Mazandaran province of Iran, a diverse range of 26 hard tick species inhabit different geographical areas, i.e., coastal plains, mountains, and forests (Nabian et al. 2007; Nasibeh et al. 2010; Rahbari et al. 2007; Razmi et al. 2007; Vahedi Noori et al. 2015). In previous sampling efforts, our focus was on the coastal plains and mountains, during which we investigated 11 tick species for the presence of Borrelia in this province (Naddaf et al. 2020). In the present study, we extended our survey to include hard ticks from forest areas of this province. We examined Borrelia infection in these hard ticks and compared their genetic identity with the species and isolates available in the GenBank database.
Materials and methods
Study area
Mazandaran province, located in the north of Iran, is characterized by its lush green landscape, situated between the Caspian Sea and north of the Alborz Mountains.
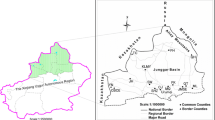
According to the Köppen-Geiger classification, the climate in this region falls under type C, subdivision Csa, a Mediterranean climate with hot summers (Kottek et al. 2006; Raziei 2017). Mazandaran province encompasses a stretch of the predominantly temperate deciduous Hyrkanian Mixed Forests belt, which extends across the northern slopes of the Alborz Mountains to the southern shores of the Caspian Sea. These ancient forests, estimated to be around 20–50 million years old, serve as vital refugia for many Arctic Tertiary relict species (Leestmans 2005; Zohary 1973) (Fig. 1).

Map showing the studied area in Mazandaran province (in grey), northern Iran
Collection and identification of ticks
Ticks were manually collected using fine stainless-steel tweezers from domestic animals, including cattle, goats, sheep, and dogs. Additionally, dragging and flagging methods were employed across the forest floor or atop vegetation in wooded areas within Amol, Babol, Savadkuh, and Sari Counties (Fig. 1). These regions are characterized by heavy rainfall and high humidity. Sampling was conducted in October 2021, during which the temperature ranged from 14 °C to 16 °C, with moisture levels from 50 to 70% in the study area. After collection, ticks were transferred into sterile 50-ml Falcon tubes, and relevant data, including location, collection date, and coordinates, were recorded and stored in sealed Styrofoam containers lined with damp cotton pads. All collected ticks were transferred to the Parasitology Laboratory of the Amol Research Center, Pasteur Institute of Iran, where they were identified based on morphological features described in taxonomic keys (Apanaskevich and Horak 2005, 2008; Apanaskevich et al. 2010; Hosseini-Chegeni 2013; Hosseini-Chegeni et al. 2019; McCoy et al. 2014; Walker 2003; Walker 2000). The specimens were grouped according to developmental host, species, developmental stage (adult, nymph, and larvae), and gender and then transferred to the Bacteriology Laboratory, Pasteur Institute of Tehran, and stored at 4 °C until DNA extraction.
DNA extraction
Following the journey, only some female ticks survived, while all males and nymphs perished. The dead specimens were transferred to 70% ethanol. Ticks were washed twice with 75% ethanol and phosphate-buffered saline (PBS), as previously described by Jiao et al. (2021). Live ticks were dissected, and their midguts were extracted and preserved in PBS at 4 °C; dead specimens could not be dissected due to desiccation. DNA was extracted from individuals or pools of midgut and whole ticks, grouped by host, species, developmental stage, and gender. Forty-one ticks were tested individually: six live females, one larva, and 34 dead ticks comprising females, males, and nymphs. The remaining specimens were grouped into pools, ranging from 2 to 40 for adults and 2 to 160 for nymphs. Samples were homogenized in 2 ml tubes containing 300–800 µL PBS and 5-mm stainless steel beads (QIAGEN, Hilden, Germany) using a homogenizer (TissueLyser II, QIAGEN). DNA extraction was then performed from 200 µL of the homogenized ticks using the potassium acetate procedure developed by Rodríguez (2014) and successfully deployed by others (Ghasemi et al. 2021; Naddaf et al. 2020). The optical density (OD) of DNA extracted from dead ticks ranged from 0.03 to 126.2 ng/µl, and from midguts ranged from 0.3 to 2620.3 ng/µl.
Real-time PCR (qPCR)
Detection of Borrelia in the whole genomic DNA extracted from ticks was conducted using primers and the probe specific for amplifying a 136-bp sequence of 16 S rRNA in the genus Borrelia (Table 1).
Results
Tick identification
In Total, 1,756 were collected by hand from 90 domestic animals, including 86 cattle, two goats, one sheep, and one dog. Additionally, 293 ticks were collected using dragging and flagging methods. Four species were identified, with Ix. ricinus being the most abundant (n = 1651), followed by Rhipicephalus annulatus (n = 384), Haemaphysalis punctata (n = 11), and Hyalomma marginatum (n = 3). Among the collected specimens, there were 319 nymphs of Ix. ricinus and 44 nymphs of Rh. annulatus, along with one Rh. annulatus larva (Table 2).
qPCR amplification
Of the 231 DNA samples, 26 pools comprising female, male, and nymphs of Rh. annulatus (n = 17) and Ix. ricinus (n = 9) ticks, along with one individual female Ha. punctata tick were positive for Borrelia DNA. The cycle thresholds (Ct) for the controls containing approximately 60’000, 6’000, 600, 60, and 6 bacteria DNA copy numbers were 24.5, 25.5, 26.5, 27.5, and 28.32, respectively. The Ct for the qPCR-positive samples ranged from 25.5 to 42.1 (Table 3). No measurable Ct values were determined for NTCs and other tick specimens
PCR amplification and sequencing
The qPCR-positive samples were further analyzed by amplifying and sequencing partial fragments of flaB and glpQ using the primers (Table 1) and amplification conditions described previously (Naddaf et al. 2020; Toledo et al. 2010). The 25 µL reactions included 2 µL (0.5–50 ng/µL) template DNA, 12.5 µL 2X Master Mix (Ampliqon, Denmark), 10 pm each of forward and reverse primers, and DDW to final volume. The flaB sequence was amplified by deploying a nested PCR assay with two sets of specific primers. The first amplification program included an initial denaturation at 95 °C for 15 min, followed by 40 cycles at 95 °C for 30 s, annealing at 50 °C for 45 s, extension at 72 °C, and a final extension at 72 °C for 7 min. For the second round of amplification, 2 µL of 1/20 dilutions from the first PCR template were used. The program for the second PCR set was the same as before, except for the annealing temperature, which was increased to 54 °C. The glpQ amplification was programmed for an initial denaturation step at 95 °C for 10 min, followed by 35 cycles at 95 °C for 30 s, 51.8 °C for 45 s, 72 °C for 1 min, and a final extension step at 72 °C for 7 min. A reaction containing all reagents except DNA was included in all assays as a negative control.
PCR products were electrophoresed on 1.5% agarose gels stained with Green Viewer (Eco DNA Safe Stain, Kala Zist, Iran) alongside a molecular weight marker and visualized under ultraviolet radiation in a gel docking device (UVITEC Cambridge Gel Documentation System, UK). Amplicons were sequenced in both directions using the Sanger method with the same primers utilized for amplification at the Sequencing Department of the Pasteur Institute, Iran, employing an ABI-3500 Sequencer. The sequences were manually checked for ambiguities by the BioEdit Sequencing Alignment Editor software (version 7.2.5), and consensus sequences were obtained. The generated sequences were deposited in the GenBank database under the accession numbers OR037292- OR037295 for glpQ and OR037296-OR037302 for flaB.
BLAST and phylogenetic analysis
The generated sequences were compared with the Borrelia sequences in the GenBank database using the Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov), and similarities were obtained. DNA sequences were aligned using Clustal W multiple alignments (Thompson et al. 1994). Then, phylogenetic analysis was performed using MEGA 11.0.10 (Tamura et al. 2021), and evolutionary flaB and glpQ trees were constructed with obtained sequences along with similar ones sourced from the GenBank database as detailed in the supplementary material (Tables S1 and S2) using the maximum likelihood algorithm based on the best-fit model (Kumar et al. 2001), i.e., Tamura 3-parameter (T92) for the flaB and Tamura-Nei (TN93) for the glpQ. Bootstrap values were calculated from 1000 replications, and the trees were rooted using the midpoint approach.
PCR amplification and sequencing
Nine qPCR-positive samples yielded the expected 604-bp flaB band, eight from Rh. annulatus and one from Ix. ricinus ticks. Also, six representatives from Rh. annulatus exhibited the 453-bp glpQ amplicon. One flaB amplicon (Ir-Maz1B1) exhibited background sequencing noises, and two glpQ amplicons (Ir-Maz 3 and Ir-Maz 1B1) yielded short sequencing results and thus were excluded from further analysis (Table 3)
BLAST analysis
Among the seven flaB sequences from Rh. annulatus ticks, five (Ir-Maz1, Ir-Maz2, Ir-Maz3, Ir-Maz1A5, and Ir-Maz1A8) were identical and matched 99.8% and 99.3% with two other sequences (Ir-Maz1A7 and Ir-Maz2C9). The flaB sequences exhibited 98.7-99.6% similarity with B. theileri C5 (acc. no. MG601737), 98.5-100% with B. theileri CATMAR-HS (acc. no. MG564190), 98.1-98.5% with B. theileri KAT (acc. no. KF569936), and 98.5-98.9% with B. theileri W27 (acc. no. LC656218). They also matched 96.2-96.6% with B. lonestari (acc. no. U26705) and 89-89.3% with B. miyamotoi Ir-Maz57 (acc. no. MN958345). The only generated flaB sequence from Ix. ricinus ticks (Ir-Maz1B11) matched 100% with B. miyamotoi (acc. no. KX646199).
The four glpQ sequences from Rh. annulatus ticks were identical and matched 98.2% with B. theileri KAT (acc. no. KF569936), 92.9% with B. lonestari MO2002-V1 (acc. no. AY682922), and 92.6% with B. miyamotoi Yekat-18 (acc. no. CP037471).
Phylogenetic analysis
In both trees, the hard tick relapsing fever (HTRF) sequences, including those generated in this study, were grouped separately from soft tick relapsing fever (STRF) borreliae and B. recurrentis. Additionally, B. miyamotoi appeared in a distant clade separate from other HTRF borreliae. In the flaB tree, our generated sequences, B. theileri C5 from Brazil, B. theileri CATMAR-HS from Egypt, B. theileri KAT from Mali, B. theileri W27 from Zambia, and undefined isolates Borrelia sp. BR and Borrelia sp. T23, from Brazil and Madagascar, formed a separate clade that branched into four subclades, close to B. lonestari and distant from Borrelia sp. RSF2354, and Borrelia sp. Ir-Maz173, previously detected in Rhipicephalus ticks in Portugal and Iran. Six generated flaB sequences emerged as a sister taxon to B. theileri C5, and one grouped with B. theileri CATMAR-HS and Borrelia sp. BR and Borrelia sp. T23 from Brazil and Madagascar. Borrelia theileri KAT from Mali and B. theileri W27 from Zambia appeared in the third and fourth subclades (Fig. 2)

The phylogenetic tree constructed based on the analyzed flaB partial sequence. The sequences generated in this study are in red and marked with asterisks. Black asterisks show sequences from Iran reflected in previous reports. Numbers at nodes refer to bootstrap probabilities; support values < 70 are not shown here. The scale bar indicates 0.02 estimated nucleotide substitutions per site. Borrelia spp. origin: R. a, Rhipicephalus annulatus; R. m, Rhipicephalus microplus; R. g, Rhipicephalus geigyi; A. m, Aepyceros melampus; A. a, Amblyomma americanum; R. s, Rhipicephalus sanguineus s.l; I. r, Ixodes ricinus; O. p, Ornithodoros parkeri; O. h, Ornithodoros hermsi; O. c, Ornithodoros coriaceus; O. t, Ornithodoros tholozani; O. e, Ornithodoros erraticus; O. s, Ornithodoros sonrai; H. s, Homo sapiens In the glpQ tree, our sequences appeared as a sister group alongside the only available sequence from B. theileri KAT (acc. no. KF569938), sharing a common ancestor with B. lonestari MO2002-V1 (acc. no. AY682922) (Fig. 3)

The phylogenetic tree based on the analyzed glpQ partial sequence. The sequences generated in this study are in red and marked with asterisks. Black asterisks show sequences from Iran reflected in previous reports. The numbers at the nodes refer to the bootstrap probabilities; support values < 70 are not shown here. The scale bar indicates 0.02 estimated nucleotide substitutions per site. Borrelia spp. origin: H.s, Homo sapiens; O. e, Ornithodoros erraticus; O. s, Ornithodoros sonrai; O.c, Ornithodoros coriaceus; O. h, Ornithodoros hermsi; A. a, Amblyomma americanum; R. g, Rhipicephalus geigyi; R. a, Rhipicephalus annulatus
Discussion
Borrelia theileri, a causative agent of bovine borreliosis, was initially identified in South Africa in Rhipicephalus ticks by Arnold Theiler over a century ago (Theiler 1904). This spirochete has since been reported in various domestic animals, including cattle, goats, sheep, and horses across Africa, North and South America, and Australia (Callow 1967; Cutler et al. 2012; Uilenberg et al. 1988). Transmission of B. theileri occurs through the infective bites of Rhipicephalus (Boophilus) ticks, such as Rhipicephalus microplus, Rh. annulatus, Rh. evertsi, and Rh. decoloratus, to domestic ruminants (Qiu et al. 2021).
This spirochete typically induces a mild febrile disease in cattle, sometimes leading to hemoglobinuria and anemia (Callow 1967). Nevertheless, B. theileri remains one of the least characterized pathogenic Borrelia transmitted by ticks (McCoy et al. 2014). Currently, no isolates of this species are available, and efforts to culture this spirochete have been unsuccessful (McCoy et al. 2014; Smith et al. 1985). Only a limited number of DNA sequences are available to distinguish this spirochete from closely related species harbored by hard ticks (McCoy et al. 2014).
In the present study, we report the presence of B. theileri in Rh. annulatus ticks from Iran for the first time. Rhipicephalus annulatus is a one-host tick, meaning once the larva hatches and finds a host, it typically develops to the next life stages on the same animal (D’Amico et al. 2017; Wang et al. 2017). We initially aimed to study Borrelia infection alongside blood sources in individual ticks. However, the low infection rate observed in early assays led us to pool the specimens. We identified B. theileri DNA in all pooled life stages of Rh. annulatus ticks collected from cattle. However, not all ticks from a single host were infected, implying the ticks might have acquired the infection during a short period of spirochetemia. Borrelia DNA was also detected in unfed adults and nymphs, suggesting transstadial transmission.
Our flaB sequences displayed the highest identity with B. theileri isolates from Brazil, Egypt, Mali, and Zambia. In phylogenetic analysis, these sequences formed a distinct clade along with B. theileri and closely related isolates from Brazil and Madagascar, further branching into four subclades (Fig. 2). Similarly, in the glpQ tree, our sequences and B. theileri from Rh. geygi in Mali diverged into two subclades (Fig. 3). This divergence suggests potential subspeciation in B. theileri, possibly driven by adaptations to different tick species infesting vertebrate hosts across diverse geographical areas. Borrelia sp. Ir-Maz173, previously identified in Rhipicephalus ticks in northern Iran (Naddaf et al. 2020), exhibited a considerable genetic similarity of 96.9-97.2% with our flaB sequences. However, in phylogenetic analysis, this isolate appeared as a sister taxon alongside an isolate from Portugal, suggesting a somewhat distant relationship from B. theileri (Fig. 2).
Our qPCR also detected Borrelia in other DNA samples obtained from Ix. ricinus and Ha. punctata tick specimens. However, despite low Ct values, we could not amplify the flaB and glpQ genes from these specimens. Previous reports have also documented the higher sensitivity of qPCR compared to conventional PCRs (Gil et al. 2005; Naddaf et al. 2020; Ornstein and Barbour 2006). Notably, DNAs extracted from the midguts of ticks (samples Ir-Maz1, IrMaz2, and Ir-Maz3) had higher Ct values in qPCR, yet yielded sharper flaB and glpQ bands and superior sequencing results (Table 3).
Various tick species infest livestock in Mazandaran province (Nabian et al. 2007; Nasibeh et al. 2010; Rahbari et al. 2007; Razmi et al. 2007; Vahedi Noori et al. 2015); however, we only captured four species, possibly due to limited sampling time and a focus on forest areas. Ixodes ricinus was the most collected species, similar to our previous survey in Mazandaran province (Naddaf et al. 2020).
Borrelia theileri DNA has also been identified in Ornithodoros ticks (Safdie et al. 2010) and human head lice (Amanzougaghene et al. 2016). Soft ticks may acquire this agent during the spirochetemia or via co-feeding transmission and subsequently test positive in PCR assays (Filatov et al. 2023). However, infection in head lice remains controversial. Head lice are very specific to humans and could have only received the agent via feeding on B. theileri-infected individuals, which makes the scenario more dilemmatic.
Iran is home to several veterinary ticks-borne infectious diseases like anaplasmosis, babesiosis, and tularemia manifested by fever, anemia, and jaundice (Esmaeili et al. 2019; Haghi et al. 2017; Hosseini-Vasoukolaei et al. 2014; Mirahmadi et al. 2022). Hence, despite displaying similar but mild symptoms (Abanda et al. 2019), screening for bovine borreliosis could offer a broader insight into circulating infectious diseases in livestock. Rhipicephalus annulatus has been documented in Lorestan, Mazandaran, Golestan, and Gilan provinces in Iran (Davari et al. 2017; Nabian et al. 2007; Ronaghi et al. 2015; Saidi et al. 2016; Telmadarraiy et al. 2010; Ziapour et al. 2017) as well as in neighboring countries such as Turkey, Iraq, and Pakistan (Ghafar et al. 2020; Jalil and Zenad 2016; Koc et al. 2015). Further investigation of ticks, livestock, and wild animals in various regions of Iran and neighboring countries can enhance our understanding of B. theileri and its relationship with other closely related species that infect Rhipicephalus ticks.
Conclusions
In this study, we have uncovered the presence of B. theileri in Rh. annulatus ticks in the north of Iran. The high identity of generated partial flaB and glpQ sequences with B. theileri, along with their clustering within well-supported clades with this species and closely related isolates from various geographical origins, offer conclusive evidence for the occurrence of B. theileri in this area. The observed divergence in B. theileri flaB and glpQ sequences from multiple geographical regions suggests potential subspeciation driven by adaptations to different tick species. This divergence in our flaB sequences implies the possible introduction of B. theileri-infected ticks from different geographical origins into Iran. As B. theileri has not been previously detected in domestic animals in Iran, monitoring this infection could enhance our understanding of febrile diseases in livestock on a broader scale.
Reactions were performed in 20 µL containing 4 µL (∼50 nM) of template DNA, 10 µL of Master Mix 2X (Amplicon, Denmark), 500 nM of each primer, 200 nM of the probe, and deionized-distilled water (DDW) to the final volume. Amplifications were performed using an ABI StepOnePlus Real-Time PCR system (Applied BioSystems, USA). The amplification protocol included an initial denaturation step at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 60 s. DDW was included as a no-template control (NTC) in all assays to ensure the reagents were free of DNA contamination. Borrelia burgdorferi sensu stricto DNA (AmpliRun® Borrelia DNA) was used in the control reactions with approximately 60,000, 6,000, 600, 60, and 6 bacteria DNA copies per reaction to determine the limit of detection, as previously described by Naddaf et al. (2020).
Data availability
No datasets were generated or analysed during the current study.
References
Abanda B, Paguem A, Abdoulmoumini M, Kingsley MT, Renz A, Eisenbarth A (2019) Molecular identification and prevalence of tick-borne pathogens in zebu and taurine cattle in North Cameroon. Parasit Vectors 12:1–3. https://doi.org/10.1186/s13071-019-3699-x
Amanzougaghene N, Akiana J, Mongo Ndombe G, Davoust B, Nsana NS, Parra HJ, Fenollar F, Raoult D, Mediannikov O (2016) Head lice of pygmies reveal the presence of relapsing fever borreliae in the Republic of Congo. PLoS Negl Trop Dis 10(12):e0005142. https://doi.org/10.1371/journal.pntd.0005142
Apanaskevich DA, Horak IG (2005) The genus Hyalomma Koch, 1844. Ii. Taxonomic status of H.(Euhyalomma) Anatolicum Koch, 1844 and H.(E.) Excavatum Koch, 1844 (acari: Ixodidae) with redescriptions of all stages. Acarina 13:181–197
Apanaskevich DA, Horak IG (2008) The genus Hyalomma Koch, 1844: V. re-evaluation of the taxonomic rank of taxa comprising the H.(Euhyalomma) marginatum Koch complex of species (acari: Ixodidae) with redescription of all parasitic stages and notes on biology. Int J Acarol 34:13–42. https://doi.org/10.1080/01647950808683704
Apanaskevich DA, Filippova NA, Horak IG (2010) The genus Hyalomma Koch, 1844. X. Redescription of all parasitic stages of H.(Euhyalomma) scupense schulze, 1919 (= H. Detritum schulze)(acari: Ixodidae) and notes on its biology. Folia Parasitologica. https://doi.org/10.14411/fp.2010.009
Callow LL (1967) Observations on tick-transmitted spirochaetes of cattle in Australia and South Africa. Br Vet J 123:492–497. https://doi.org/10.1016/S0007-1935(17)39704-X
Cutler S, Abdissa A, Adamu H, Tolosa T, Gashaw A (2012) Borrelia in Ethiopian ticks. Ticks Tick Borne Dis 3:14–17. https://doi.org/10.1016/j.ttbdis.2011.08.004
Cutler SJ, Ruzic-Sabljic E, Potkonjak A (2017) Emerging borreliae– expanding beyond Lyme borreliosis. Mol Cell Probes 31:22–27. https://doi.org/10.1016/j.mcp.2016.08.003
D’Amico G, Mihalca AD, Estrada-Peña A (2017) Rhipicephalus annulatus (say, 1821)(Figs. 133–135). Ticks Eur N Afr: A Guide to Species Identification: 335–342
Davari B, Alam FN, Nasirian H, Nazari M, Abdigoudarzi M, Salehzadeh A (2017) Seasonal distribution and faunistic of ticks in the Alashtar County (Lorestan Province), Iran. Pan Afr Med J 27. https://doi.org/10.11604/pamj.2017.27.284.10341
Esmaeili S, Ghasemi A, Naserifar R, Jalilian A, Molaeipoor L, Maurin M, Mostafavi E (2019) Epidemiological survey of tularemia in Ilam Province, West of Iran. BMC Infect Dis 19:502. https://doi.org/10.1186/s12879-019-4121-1
Filatov S, Krishnavajhala A, Lopez JE (2023) Autogenous reproduction by Ornithodoros turicata (ixodida: Argasidae) females and vertical transmission of the tick-borne pathogen Borrelia turicatae (spirochaetales: borreliaceae). Appl Environ Microbiol 89(11):e01032–e01023. https://doi.org/10.1128/aem.01032-23
Furuno K, Lee K, Itoh Y, Suzuki K, Yonemitsu K, Kuwata R, Shimoda H, Watarai M, Maeda K, Takano A (2017) Epidemiological study of relapsing fever borreliae detected in Haemaphysalis ticks and wild animals in the western part of Japan. PLoS ONE 12(3):e0174727. https://doi.org/10.1371/journal.pone.0174727
Ghafar A, Gasser RB, Rashid I, Ghafoor A, Jabbar A (2020) Exploring the prevalence and diversity of bovine ticks in five agro-ecological zones of Pakistan using phenetic and genetic tools. Ticks Tick Borne Dis 11:101472. https://doi.org/10.1016/j.ttbdis.2020.101472
Ghasemi A, Naddaf SR, Mahmoudi A, Rohani M, Naeimi S, Mordadi A, Cutler SJ, Mostafavi E (2021) Borrelia duttonii-like spirochetes parasitize Meriones persicus in East Azerbaijan province of Iran. Ticks Tick Borne Dis 12:101825. https://doi.org/10.1016/j.ttbdis.2021.101825
Gil H, Barral M, Escudero R, García-Pérez AL, Anda P (2005) Identification of a new Borrelia species among small mammals in areas of northern Spain where Lyme disease is endemic. Appl Environ Microbiol 71:1336–1345. https://doi.org/10.1128/aem.71.3.1336-1345.2005
Haghi MM, Etemadifar F, Fakhar M, Teshnizi SH, Soosaraei M, Shokri A, Hajihasani A, Mashhadi H (2017) Status of babesiosis among domestic herbivores in Iran: a systematic review and meta-analysis. Parasitol Res 116:1101–1109
Hosseini-Chegeni A, Hosseini R, Tavakoli M, Telmadarraiy Z, Abdigoudarzi M (2013) The Iranian Hyalomma, with a key to the identification of male species. Persian J Acarol 2:503–529. https://doi.org/10.22073/pja.v2i3.10046
Hosseini-Chegeni A, Tavakoli M, Telmadarraiy Z (2019) The updated list of ticks (acari: Ixodidae & argasidae) occurring in Iran with a key to the identification of species. Syst Appl Acarol 24:2133–2166. https://doi.org/10.11158/saa.24.11.8
Hosseini-Vasoukolaei N, Oshaghi MA, Shayan P, Vatandoost H, Babamahmoudi F, Yaghoobi-Ershadi MR, Telmadarraiy Z, Mohtarami F (2014) Anaplasma infection in ticks, livestock and human in Ghaemshahr, Mazandaran Province, Iran. J Arthropod Borne Dis 8:204
Houmansadr F, Soleimani M, Naddaf SR (2020) Development of a loop-mediated isothermal amplification (lamp) assay for detection of relapsing fever borreliae. J Arthropod Borne Dis 14:47. https://doi.org/10.18502/jad.v14i1.2703
Jalil WI, Zenad MM (2016) Isolation of aerobic bacteria from ticks infested sheep in Iraq. Asian Pac J Trop Biomed 6:67–70
Jiao J, Lu Z, Yu Y, Ou Y, Fu M, Zhao Y, Wu N, Zhao M, Liu Y, Sun Y, Wen B (2021) Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasit Vectors 14:287. https://doi.org/10.1186/s13071-021-04740-3
Koc S, Aydın L, Cetin H (2015) Tick species (acari: Ixodida) in Antalya city, Turkey: species diversity and seasonal activity. Parasitol res 114:2581–2586
Kottek M, Grieser J, Beck C, Rudolf B, Rubel F (2006) World map of the köppen-geiger climate classification updated. Meteorol Z. https://doi.org/10.1127/0941-2948/2006/0130
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) Mega2: molecular evolutionary genetics analysis software. Bioinform 17:1244–1245. https://doi.org/10.1093/bioinformatics/17.12.1244
Leestmans R (2005) Le refuge Caspiens Et son importance en biogéographie. Linneana Belg 20:97–102
Marti Ras N, Lascola B, Postic D, Cutler SJ, Rodhain F, Baranton G, Raoult D (1996) Phylogenesis of relapsing fever Borrelia spp. Int J Syst Evol Microbiol 46:859–865. https://doi.org/10.1099/00207713-46-4-859
McCoy BN, Maïga O, Schwan TG (2014) Detection of Borrelia theileri in Rhipicephalus geigyi from Mali. Ticks Tick Borne Dis 5:401-3. https://doi.org/10.1016/j.ttbdis.2014.01.007
Mirahmadi H, Ghaderi A, Barani S, Alijani E, Mehravaran A, Shafiei R (2022) Prevalence of camel babesiosis in southeast of Iran. Vet Med Sci 8:343–348. https://doi.org/10.1002/vms3.666
Nabian S, Rahbari S, Shayan P, Hadadzadeh HR (2007) Current status of tick fauna in north of Iran. Iran J Parasitol 2:12–17
Naddaf SR, Ghazinezhad B, Bahramali G, Cutler SJ (2012) Phylogenetic analysis of the spirochete Borrelia microti, a potential agent of relapsing fever in Iran. J Clin Microbiol 50:2873–2876. https://doi.org/10.1128/jcm.00801-12
Naddaf SR, Ghazinezhad B, Sedaghat MM, Asl HM, Cutler SJ (2015) Tickborne relapsing fever in southern Iran, 2011–2013. Emerg Infect Dis 21:1078–1080. https://doi.org/10.3201/eid2106.141715
Naddaf SR, Ghazinezhad B, Kazemirad E, Cutler SJ (2017) Relapsing fever causative agent in Southern Iran is a closely related species to east African borreliae. Ticks Tick Borne Dis 8:882–886. https://doi.org/10.1016/j.ttbdis.2017.07.006
Naddaf SR, Mahmoudi A, Ghasemi A, Rohani M, Mohammadi A, Ziapour SP, Nemati AH, Mostafavi E (2020) Infection of hard ticks in the Caspian Sea littoral of Iran with Lyme borreliosis and relapsing fever borreliae. Ticks Tick Borne Dis 11:101500. https://doi.org/10.1016/j.ttbdis.2020.101500
Nasibeh HV, Zakkyeh T, Hassan V, Reza YE, Morteza HV, Ali OM (2010) Survey of tick species parasiting domestic ruminants in Ghaemshahr County, Mazandaran Province, Iran. Asian Pac J Trop Med 3:804–806. https://doi.org/10.1016/S1995-7645(10)60193-9
Ornstein K, Barbour AG (2006) A reverse transcriptase-polymerase chain reaction assay of Borrelia burgdorferi 16S rRNA for highly sensitive quantification of pathogen load in a vector. Vector Borne Zoonotic Dis (Larchmont NY) 6:103–112. https://doi.org/10.1089/vbz.2006.6.103
Qiu Y, Squarre D, Nakamura Y, Lau AC, Moonga LC, Kawai N, Ohnuma A, Hayashida K, Nakao R, Yamagishi J, Sawa H, Namangala B, Kawabata H (2021) Evidence of Borrelia theileri in wild and domestic animals in the Kafue ecosystem of Zambia. Microorganisms 9:2405. https://doi.org/10.3390/microorganisms9112405
Rahbari S, Nabian S, Shayan P (2007) Primary report on distribution of tick fauna in Iran. Parasitol Res 101:175–177. https://doi.org/10.1007/s00436-007-0692-7
Raziei T (2017) Köppen-geiger climate classification of Iran and investigation of its changes during 20th century. J Earth Space Phys 43:419–439. https://doi.org/10.22059/jesphys.2017.58916
Razmi GR, Glinsharifodini M, Sarvi S (2007) Prevalence of ixodid ticks on cattle in Mazandaran Province, Iran. Korean J Parasitol 45:307
Rodríguez I, Fraga J, Noda AA, Mayet M, Duarte Y, Echevarria E, Fernández C (2014) An alternative and rapid method for the extraction of nucleic acids from Ixodid ticks by potassium acetate procedure. Braz Arch Biol Technol 57:542–547. https://doi.org/10.1590/S1982-88372014000100011
Ronaghi H, Nabian S, Ebrahimzadeh E, Biranvand F, Shayan P (2015) Molecular characterization of Rhipicephalus (Boophilus) annulatus from Iran by sequences of cytochrome c oxidase subunit I (COI) and the second internal transcribed spacer (its2). Iran J Vet Med 9:117–123. https://doi.org/10.22059/ijvm.2015.54010
Safdie G, Farrah IY, Yahia R, Marva E, Wilamowski A, Sawalha SS, Wald N, Schmiedel J, Moter A, Göbel UB, Bercovier H, Abdeen Z, Assous MV, Fishman Y (2010) Molecular characterization of Borrelia persica, the agent of tick borne relapsing fever in Israel and the Palestinian authority. PLoS ONE 5:e14105. https://doi.org/10.1371/journal.pone.0014105
Saidi S, Nabian S, Ebrahimzade E, Najafi A, Moosazadeh Moghaddam M, Sazmand A, Torkzadeh-Mahani M, Tabrizi SS (2016) Identification and characterization of a cathepsin l-like cysteine protease from Rhipicephalus (Boophilus) annulatus. Exp Appl Acarol 68:251–265. https://doi.org/10.1007/s10493-015-9993-1
Shirani D, Rakhshanpoor A, Cutler SJ, Ghazinezhad B, Naddaf SR (2016) A case of canine borreliosis in Iran caused by Borrelia persica. Ticks Tick Borne Dis 7:424–426. https://doi.org/10.1016/j.ttbdis.2015.12.020
Smith RD, Miranpuri GS, Adams JH, Ahrens EH (1985) Borrelia theileri: isolation from ticks (Boophilus microplus) and tick-borne transmission between splenectomized calves. Am J Vet Res 46:1396–1398
Talagrand-Reboul E, Boyer PH, Bergström S, Vial L, Boulanger N (2018) Relapsing fevers: neglected tick-borne diseases. Front Cell Infect Microbiol 8:98. https://doi.org/10.3389/fcimb.2018.00098
Tamura K, Stecher G, Kumar S (2021) Mega11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Telmadarraiy Z, Ghiasi SM, Moradi M, Vatandoost H, Eshraghian MR, Faghihi F, Zarei Z, Haeri A, Chinikar SA (2010) A survey of Crimean-Congo haemorrhagic fever in livestock and ticks in Ardabil province, Iran during 2004–2005. Scand J Infect Dis 42:137–141. https://doi.org/10.3109/00365540903362501
Theiler A (1904) Spirillosis of cattle. J Clin Pharm Ther 17:47–55. https://doi.org/10.1016/S0368-1742(04)80003-1
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. https://doi.org/10.1093/nar/22.22.4673
Toledo A, Anda P, Escudero R, Larsson C, Bergstrom S, Benach JL (2010) Phylogenetic analysis of a virulent Borrelia species isolated from patients with relapsing fever. J Clin Microbiol 48:2484–2489. https://doi.org/10.1128/jcm.00541-10
Trevisan G, Cinco M, Trevisini S, di Meo N, Chersi K, Ruscio M, Forgione P, Bonin S (2021) Borreliae part 1: Borrelia lyme group and Echidna-Reptile group. Biology 10:1036
Uilenberg G, Hinaidy HK, Perié NM, Feenstra T (1988) Borrelia infections of ruminants in Europe. Vet Q 10:63–67. https://doi.org/10.1080/01652176.1988.9694148
Vahedi Noori N, Abdi Goodarzi M, Kiasari MN (2015) Evaluation of the species diversity and abundance of hard ticks (family: Ixodidae) parasite of cattle and sheep in Mazandaran Province. Vet Res Biol Prod 28:58–64. https://doi.org/10.22092/vj.2015.100915
Walker JB, Keirans JE, Horak IG (2000) The genus Rhipicephalus (acari, ixodidae): a guide to the brown ticks of the world. Cambridge univ press http://https://doi.org/10.1017/CBO9780511661754
Walker AR, Bouattour A, Camicas JL, Estrada-Pena A, Horak IG, Latif A, Pegram RG, Preston PM (2003) Ticks of domestic animals in Africa: a guide to identification of species. Edinb Biosci Rep 74
Wang HH, Corson MS, Grant WE, Teel PD (2017) Quantitative models of Rhipicephalus (Boophilus) ticks: historical review and synthesis. Ecosphere 8:e01942
Wodecka B (2007) Significance of red deer (Cervus elaphus) in the ecology of Borrelia burgdorferi sensu lato. Wiad Parazytol 53:231–237
Ziapour SP, Kheiri S, Fazeli-Dinan M, Sahraei-Rostami F, Mohammadpour RA, Aarabi M, Nikookar SH, Sarafrazi M, Asgarian F, Enayati A, Hemingway J (2017) Pyrethroid resistance in Iranian field populations of Rhipicephalus (Boophilus) annulatus. Pestic Biochem Physiol 136:70–79. https://doi.org/10.1016/j.pestbp.2016.08.001
Zohary M (1973) Geobotanical foundations of the Middle East. Gustav Fischer Verlag, Stuttgart, Swets & Zeitlinger, Amsterdam
Acknowledgements
Acknowledgments We thank The Pasteur Institute of Iran for funding this study. We also thank Shadi Aghamohammad and Sepideh Fereshteh for assisting in setting up the qPCR and dissecting the ticks.
Funding
Pasteur Institute of Iran Funded this study (Grant No.: 1250).
Author information
Authors and Affiliations
Contributions
SRN and MR conceived and designed the study; SPZ and SRN participated in sample collection and identification; MM, SRN, MR, and AAS performed laboratory experiments and analyzed data. SRN, MM, MR, and SPZ contributed to writing and revising the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The ethical committee of the Pasteur Institute of Iran approved this project (approval ID: IR.PII. REC.1400.051).
Consent for publication
All authors who participated in the study concurred with the submission and subsequent revisions submitted by the corresponding authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Milani, M., Naddaf, S.R., Ziapour, S.P. et al. Borrelia theileri infections in Rhipicephalus annulatus ticks from the north of Iran. Exp Appl Acarol 93, 81–95 (2024). https://doi.org/10.1007/s10493-024-00924-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-024-00924-5