
Abstract
Haemaphysalis longicornis is an ixodid tick that can spread a wide variety of pathogens, affecting humans, livestock and wildlife health. The high reproductive capability of this species is initiated by the ingestion of a large amount of blood ingested by the engorged female tick. The degree of ovarian development is proportional to the number of eggs laid. Studying the regulatory mechanism of tick ovary development is relevant for the development of novel tick control methods. In this study, we used quantitative proteomics to study the dynamic changes in protein expression and protein phosphorylation during ovarian development of engorged female H. longicornis ticks. Synergistic action of many proteins (n = 3031) is required to achieve ovarian development and oocyte formation rapidly. Through bioinformatics analysis, changes in protein expressions and phosphorylation modifications in regulating the ovarian development of female ticks are described. Many proteins play an essential role during ovarian development. Also, protein phosphorylation appeared an important reproductive strategy to enable ticks to efficiently convert large amounts of blood in the ovaries into egg-producing components and ultimately produce many eggs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Ticks are obligate hematophagous ectoparasites of all terrestrial vertebrates, are second only to mosquitoes as vectors of human pathogens (Bartíková et al. 2017). They are widely distributed throughout the world and parasitize mammals, birds, reptiles, and a small number of amphibians. Ticks carry many pathogens, which are transmitted to the host during blood feeding, causing great harm to human health, livestock production, and the reproduction of wildlife populations (Cayol et al. 2017).
Haemaphysalis longicornis belongs to the family Ixodidae. It is mainly distributed in eastern Asia and Australia and transmits a variety of pathogens (Otaki et al. 2018), such as viruses, bacteria, rickettsia, spirochetes, and protozoa (de la Fuente 2018; Wada et al. 2010). Ticks are extremely harmful blood-feeding animals. The more ticks there are, the greater the harm they cause. The degree of ovarian development directly affects the number of eggs laid by the female tick, the survival rate of the eggs, the hatching rate, and the size of the tick population. After engorgement, the ovary enters a rapid developmental stage (Ullah and Kaufman 2014), the axial diameter of the oocyte is rapidly enlarged, and the rapidly enlarged oocyte protrudes from the ovary wall (Yang et al. 2014), in which it matures and rapidly produces eggs under the synergistic action of many proteins. Currently, it is believed that during embryo maturation, vitellogenin (Vg) is synthesized, released into the haemolymph, selectively taken up and then processed by developing oocytes to form vitelline (Vn) as a nutritional source of embryogenesis (Raikhel and Dhadialla 1992). However, the specific regulatory mechanism of Vg processing into Vn, as well as other related regulatory mechanisms affecting the rapid ovary development, remains unclear (Xavier et al. 2018). Many proteins are involved in these regulatory mechanisms, which can be elucidated by investigating the changes in the expression levels of these proteins and their post-translational modifications. Currently, proteomics technology is the most powerful method to quickly understand the mechanism of these protein changes. Some previous studies have used proteomics to study changes in proteins during ovarian development of ticks (Xavier et al. 2018; Rachinsky et al. 2007; Ramírez Rodríguez et al. 2016). However, with the continuous innovation of protein quantification technology, novel research methods can more accurately and clearly explore the protein dynamic changes at various stages of ovarian development. In addition, various post-translational modifications of proteins can be identified and quantified by using current advanced quantitative techniques. As far as we know, studies on phosphorylated proteomics in the developmental stages of tick ovary have not yet been reported.
Data independent acquisition (DIA) is one of the most advanced quantitative proteomics approaches. The DIA method is highly sensitive and stable and is capable of simultaneously identifying different sets of samples (Ludwig et al. 2018; Rosenberger et al. 2017). It does not require labelling with stable isotopes and has thus been widely applied in proteomics studies. In this study, we adopted quantitative proteomics based on DIA to investigate dynamic changes in protein expression and phosphorylation levels during ovarian development of engorged females of H. longicornis. A total of 2803 proteins were identified, of which 506 were differentially expressed, including a variety of proteins involved in the degradation of Vn and cell proliferation. The results of the phosphoproteomics study on the ovarian development process showed that phosphorylation occurred at 1234 amino acid sites of 549 phosphorylated proteins, including proteins that regulate growth and development. This study may help identify key proteins that regulate ovarian development in ticks and provide new ideas for tick control.
Methods
Tick feeding
Haemaphysalis longicornis were collected from Zhangjiakou in Hebei Province, China. Ticks were allowed to feed on the ears of New Zealand white rabbits by using earmuffs made of white cloth. Each rabbit was kept in constant temperature culture room (25 °C). During the non-feeding period, the ticks were maintained in constant temperature (25 ± 1 °C) and humidity (75%) artificial climate incubators (Wang et al. 2018). The ticks used in this study have been cultured for multiple generations under laboratory conditions. All experimental procedures were approved by the Animal Ethics Committee of Hebei Normal University (Protocol Number: 165031).
Ovarian dissect and protein extraction
The time of natural detachment of engorged female ticks (approximately post-feeding 5.5 days, 249.2 ± 6.8 mg) from the host was recorded as 0 h. Ovaries from female ticks at the 12 h, 24 h, 36 h, and 48 h post-engorgement stages were dissected. Then, the ovaries were immediately placed in sterile PBS (0.01 M) containing NaF (0.05 M), Na3VO4 (0.01 M), and protease inhibitor cocktail (0.01 M) (Sigma, USA). The protein extraction method was the same as previously described (Hawkins et al. 2019). The extracted proteins were lyophilized and frozen at – 80 °C for subsequent experiments. Four biological replicates were performed in data independent acquisition (DIA) quantitative experiment. About 100 engorged female ticks were dissected for each stage analysis.
Protein digestion
First, 10 mM dithiothreitol was added to each protein sample (3 mg) and incubated at 37 °C for 1 h. Then, 20 mM iodoacetamide was added to each protein sample and incubated at 26 °C for 30 min in the dark. Trypsin (1:100 w/w, Thermo Fisher Scientific, USA) digestion occurred at 37 °C for 12 h. LC–MS (Thermo Fisher Scientific) was used to monitor digestion efficiency. Then, according to the manufacturer’s protocol, the digested peptides were desalinated using C18 SPE (CNW, UK). Then, the purified peptide samples were normalized to a common concentration using the BCA Protein Assay Kit (Pierce Biotechnology, USA).
Phosphorylated peptide enrichment
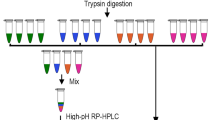
The workflow for phosphorylated peptide enrichment is shown in Fig. 1. The TiO2 beads (GL Sciences, Japan) were used to enrich the phosphorylated peptides. Aliquots of TiO2 beads were washed three times using buffer with 50% acetonitrile (ACN) containing 2% trifluoroacetic acid (TFA), saturated by glutamic acid. The TiO2 beads and peptides were dissolved in the same 800 µl buffer with gentle shaking at room temperature for 1 h. The TiO2 beads were then washed with 50% ACN to remove the non-phosphorylated peptide. Then, TiO2 beads were washed twice with 50% ACN containing 20 mM ammonium acetate. Phosphorylated peptides were then eluted from the TiO2 beads with 200 µl of 0.3 M NH4OH one time and two times with 200 µl of 0.5 M NH4OH. The enriched phosphorylated peptides were then centrifuged, concentrated, dried, and frozen at − 20 °C until they were used.

Workflow for DIA-based proteomics analysis of global proteins and phosphorylated proteins of ovarian development in female Haemaphysalis longicornis
High-pH reverse separation for data dependent acquisition (DDA) analysis and generation of spectral library
The peptides or phosphorylated peptides samples of each group were mixed in equal proportions and then separated by high-performance liquid chromatography (HPLC) (Waters e2695, USA) with Durashel-C18 column (5 µm particle size, 100 Å pore size, 4.6 × 250 mm, Agela, China), respectively. The elution solvent used solvent A (water with 5 mM ammonium formate, pH 10.0) and solvent B (ACN with 5 mM ammonium formate, pH 10.0). The flow rate was set at 1 ml/min. Elution line gradient was set at a concentration of 5% of solvent B at 15 min and stopped at a concentration of 50% of solvent B at 75 min. Sixty elutions were collected in each tube at every 1 min interval, and these elutions were then combined into 10 groups for further LC–MS analysis to construct a spectral library.
Spectral image library construction
To each group peptide sample, the indexed Retention Time (iRT) reagent (Biognosys, Switzerland) was added. Then, the sample was analysed using an M-Class nanoACQUITY ultra performance liquid chromatography (UPLC) (Waters, USA) and a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Each sample was analysed by a C18 RP analytical column (1.8 µm particle size, 100 µm × 150 mm; Waters, USA). The UPLC elution flow rate was set at 300 nl/min; the mobile phase A was water with 0.1% formic acid (FA), and the mobile phase B was ACN with 0.1% FA. The elution linear gradient was set as follows: 2–8% gradient of solvent B for 8 min, then 8–35% solvent B gradient for 117 min. The parameter settings of mass spectrometry Q Exactive HF identification were consistent with our previous reports (Hawkins et al. 2019).
Proteome Discoverer 2.2 (Thermo Fisher Scientific) was used to calculate DDA mass spectra results to construct the spectral image library. The raw data of the above 10 groups were merged into one search. The database used for DDA mass spectra searching came from the H. longicornis proteins database derived from transcriptome sequencing (NCBI Accession Number: GHLT00000000). In order to eliminate the contamination, host Oryctolagus cuniculus and human sequences were used as contaminated database for proteomic searching. The calculation parameters were as follows: HCD fracture and precursor and fragment mass tolerance of 10 ppm and 0.02 Da, respectively; trypsin digestion with 2 missed sites; variable modifications set methionine oxidation, N-terminal acetylation, phosphorylation of serine, threonine, and tyrosine; fixed modifications set carbamidomethylation of cysteine; target false discovery rate (FDR) = 0.01.
DIA identification and quantification
DIA identification was performed on each sample. The chromatographic conditions of nanoUPLC were the same as those for DDA. DIA data were analysed using Spectronaut™ software (v.11.0, Biognosys). DIA mass spectrometry parameters were set as previously described (Hawkins et al. 2019). The FDR was set to less than 1% and Q value < 0.05; other parameters were set to default values.
Bioinformatics analysis of differentially expressed proteins
To further reveal biological functions, some bioinformatics methods were used. The GProX was used to cluster the proteins with similar expression change characteristics (Rigbolt et al. 2011). The number of clusters was set to 4, and a fixed regulation threshold (upper limit of 0.58 for protein up-regulation and lower limit of – 0.58 for protein down-regulation, corresponding to the original ratios 1.5 and 0.67) was used. Principal Component Analysis (PCA) was performed with online analysis software (https://www.omicsolution.org/wu-kong-beta-linux/main/). The Gene Ontology (GO) functional annotation of the proteins was performed using the online PANTHER software (https://www.pantherdb.org/). The pathways related to differentially expressed proteins were analysed using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/kegg/).
Results
Protein identification
The numbers of identified proteins at 0, 12, 24, 36, and 48 h after engorgement were 2937, 2883, 2866, 2797, and 2480, respectively, of which the numbers of identified proteins with a coefficient of variation (CV) less than 20% among four replicates in each group were 2148, 2113, 2042, 1966, and 1715, respectively (Fig. 2a). We performed a PCA on the experimental data of the four replicates in the five stages (Fig. 3) and found that there were significant differences among the five sets of data, but not among the replicates, indicating the high reproducibility of the repeated experiments of each group. The Venn diagram analysis of the identification results (Fig. 2b) showed that the number of proteins identified in all five stages was 1158, and the information on the overlapping set is shown in Supplementary Table S1.

Statistics for the identified proteins information in the ovaries of female Haemaphysalis longicornis. a The number of proteins at five stages of post-engorgement. b Venn diagram analysis of proteins (CV < 20%) among the five stages

Principal component analysis of identified proteins in the ovaries of female Haemaphysalis longicornis at five stages of post-engorgement
The numbers of identified phosphorylated peptides at 0, 12, 24, 36, and 48 h after engorgement were 3149, 3028, 3096, 3090, and 2583, respectively, of which the numbers of the identified phosphorylated peptides with a CV value less than 20% among four replicates in each group were 2162, 2442, 2305, 2398 and 1547, respectively (Fig. 4a). The Venn diagram (Fig. 4b) shows that 1131 phosphorylated peptides were present in all five stages, in which 1234 phosphorylation sites were present. Among the peptides, 1031 peptides contained one phosphorylation site, 97 peptides contained two phosphorylation sites, and 3 peptides contained three phosphorylation sites (Fig. 4c). Among the 1234 phosphorylation sites, 83.7% occurred at the serine residue, 14.1% at the threonine residue, and 2.2% at the tyrosine residue (Fig. 4d). When the changes in the expression of the same polypeptide in different stages were more than 1.5 fold, the polypeptide was considered differentially expressed. In this study, we found that 1125 phosphorylated peptides, corresponding to 545 proteins, were differentially expressed. 344 phosphorylated peptides, corresponding to 204 phosphorylated proteins, were up-regulated, whereas 222 phosphorylated peptides, corresponding to 140 phosphorylated proteins, were down-regulated. The details are shown in Supplementary Table S2.

Statistics for the identified phosphorylated proteins information in the ovaries of female Haemaphysalis longicornis. a The number of phosphorylated peptides at five stages of post-engorgement. b Venn diagram analysis of phosphorylated peptides (CV < 20%) among the five stages. c Number of phosphorylation sites of the peptides. d Distribution of phosphorylated amino acids in serine, threonine, and tyrosine
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository. Project Name: Quantitative and phosphorylated proteomics data on the dynamic changes of ovarian developmental proteins in female Haemaphysalis longicornis. Project accession: IPX0001747000/PXD015267.
Clustering analysis on all proteins
All proteins in the stages of the 12 h/0 h, 24 h/0 h, 36 h/0 h, and 48 h/0 h that had annotation information were subjected to Cluster analysis (Fig. 5), in which the log2 values lower than – 0.58 or greater than 0.58, respectively, represented down-regulated or up-regulated protein expression. Cluster 0 included 652 proteins that were not significantly differentially expressed. Proteins with similar changing trends in expression during ovarian development were classified into four Clusters. Cluster 1 included 107 proteins whose expression assumed down-regulated trend in the mid and late stages of ovarian development. Cluster 2 included 138 proteins whose expression levels assumed a down-regulated trend during ovarian development. Cluster 3 included 195 proteins whose expression assumed an up-regulated trend during the ovarian development. Cluster 4 included 66 proteins that exhibited complicated trends and did not show a certain pattern, most of which were proteins regulating the physiological changes in cells and thus changed dynamically all the time.

Cluster analysis of differentially expressed proteins in the ovaries of female Haemaphysalis longicornis
The expression levels of proteins in Cluster 3 kept increasing during the period from 0 to 48 h post-engorgement, and the functions of the proteins include the following categories: (1) Vn; (2) protein-degrading enzymes, such as aspartic protease, cysteine protease, yolk cathepsin, and serine carboxypeptidase; (3) cell proliferation-associated proteins, such as fascin and transferrin receptor; (4) metabolism-associated proteins, such as transketolase and hexokinase; (5) cell structure and cytoskeleton, such as cortactin and erythrocyte band 7 integral membrane protein; (6) proteins that promote the metabolism of cells, such as tyrosine aminotransferase and adenosine kinase; (7) proteins with unknown functions.
GO enrichment analysis of proteins in each Cluster
The GO annotations of the differentially expressed proteins from each Cluster were shown in Fig. 6. These proteins were categorized in three categories (Molecular Function, Biological Process, and Cellular Component). The expression levels of proteins of Cluster 1 were down-regulated trend in the mid and late stages of ovarian development. In the Molecular Function category, the proteins in Cluster 1 were enriched in 6 nodes, of which the binding (45.8%) and structural molecule activity (31.3%) nodes comprised a large proportion of the proteins. In the Biological Process category, 6 nodes were enriched, of which the cellular process (35.1%) and metabolic process (27.0%) nodes accounted for a large proportion of the proteins. In the Cellular Component category, 3 nodes were enriched, the cell (67.5%) and protein-containing complex (20.0%) nodes accounted for a large proportion of the proteins.

GO functional annotations for differentially expressed proteins in four Clusters. a–d GO functional annotations for proteins in Clusters 1 to 4
The expression levels of proteins of Cluster 2 showed a down-regulated trend during ovarian development. In the Molecular Function category, the proteins in Cluster 2 were enriched in 6 nodes, of which the binding (40.4%) and catalytic activity (38.5%) nodes comprised a large proportion of the proteins. In the Biological Process category, 6 nodes were enriched, of which the metabolic process (35.8%) and localization (30.2%) nodes accounted for a large proportion of the proteins. In the Cellular Component category, 4 nodes were enriched, the cell (49.2%) and organelle (30.2%) nodes accounted for a large proportion of the proteins.
Proteins in Cluster 3 showed up-regulated trend during the ovarian development. In the Molecular Function category, the proteins in Cluster 3 were enriched in 7 nodes, of which the catalytic activity (63.3%) and binding (25.0%) nodes comprised a large proportion of the proteins. In the Biological Process category, 5 nodes were enriched, of which the metabolic process (42.9%) and cellular process (36.5%) nodes accounted for a large proportion of the proteins. In the Cellular Component category, 4 nodes were enriched, the cell (64.1%) and organelle (17.9%) nodes accounted for a large proportion of the proteins.
The expression levels of proteins of Cluster 4 were not show a certain pattern during the ovarian development. In the Molecular Function category, the proteins in Cluster 4 were enriched in 3 nodes, of which the catalytic activity binding (54.5%) and catalytic activity (36.4%) nodes comprised a large proportion of the proteins. In the Biological Process category, 6 nodes were enriched, of which the cellular process (50.0%) and localization (18.8%) nodes accounted for a large proportion of the proteins. In the Cellular Component category, 6 nodes were enriched, the cell (30.0%) and organelle (30.0%) nodes accounted for a large proportion of the proteins.
GO enrichment analysis of differentially expressed total proteins and phosphorylated proteins
GO “Protein Class” enrichment analysis was carried out on the differentially expressed 506 proteins which were identified in ovary during all five post-engorgement stages (Supplementary Fig. S1a). Most of these proteins belong to nucleic acid binding, hydrolase, enzyme modulator, oxidoreductase, transferase, and cytoskeletal protein. It was assigned them to three categories: Molecular Function, Biological Process, and Cellular Component (Supplementary Fig. S2). In the Molecular Function category, 8 nodes were enriched, of which the catalytic activity (40.9%) and binding (37.4%) nodes accounted for a large proportion of the proteins. In the Biological Process category, 7 nodes were enriched, of which the metabolic process (34.3%) and cellular processes (33.7%) nodes accounted for a large proportion of proteins. In the Cellular Component category, 6 nodes were enriched, of which the cell (54.9%) and organelle (22.8%) nodes accounted for a large proportion of the proteins.
We also performed the GO “Protein Class” enrichment analysis on the 1125 phosphorylated peptides corresponding to differentially expressed 545 proteins which were identified in ovary during all five post-engorgement stages (Supplementary Fig. S1b). Most of these proteins belonged to nucleic acid binding, enzyme modulator, hydrolase, transferase, cytoskeletal protein, and transcription factor. It was assigned them to three categories: Molecular Function, Biological Process, and Cellular Component (Supplementary Fig. S3). In the Molecular Function category, 8 nodes were enriched, of which the binding (44.2%) and catalytic activity (33.9%) nodes accounted for a large proportion of the proteins. In the Biological Process category, 11 nodes were enriched, of which the cellular processes (35.7%) and metabolic process (27.1%) nodes accounted for a large proportion of proteins. In the Cellular Component category, 6 nodes were enriched, of which the cell (42.7%) and organelle (37.6%) nodes accounted for a large proportion of proteins.
GO enrichment analysis was also performed on the 204 up-regulated proteins corresponding to the 344 phosphorylated peptides (Fig. 7a), and found that in the Molecular Function category, 7 nodes were enriched, of which the binding (38.8%) and catalytic activity (37.6%) nodes accounted for a large proportion of the proteins. Proteins related to catalytic activity included a large number of hydrolases. In the Biological Process category, 9 nodes were enriched, of which the cellular processes (39.3%) and metabolic process (23.6%) nodes accounted for a large proportion of proteins; in the Cellular Component category, 5 nodes were enriched, of which the cell (44.8%) and organelle (43.3%) nodes accounted for a large proportion of proteins.

GO functional annotations of differentially expressed phosphorylated proteins. a Up-regulated phosphorylated proteins. b Down-regulated phosphorylated proteins
Meanwhile, 140 down-regulated proteins corresponding to 222 phosphorylated peptides were carried out GO enrichment analysis (Fig. 7b). It was found that in the Molecular Function category, 4 nodes were enriched, of which the binding (55.2%) and catalytic activity (29.9%) nodes accounted for a large proportion of the proteins. Proteins related to catalytic activity included a large number of hydrolases. In the Biological Process category, 9 nodes were enriched, of which the metabolic process (37.2%) and cellular processes (26.9%) nodes accounted for a large proportion of proteins; in the Cellular Component category, 4 nodes were enriched, of which the organelle (40.7%) and cell part (37.0%) nodes accounted for a large proportion of proteins.
KEGG pathway analysis of differentially expressed proteins
We performed KEGG pathway analysis on differentially expressed 506 proteins and found that they were involved in 158 pathways. The top 10 pathways in which these proteins were involved (Fig. 8). 74 proteins were involved in metabolic pathways, and the continuous changes in the expression levels of these proteins indicate that some metabolic processes in ovarian development also changed from time to time. 21 proteins were involved in ribosome pathway and might be closely related to protein synthesis during ovarian development.

The top 10 pathways of KEGG pathway enrichment analysis of differentially expressed proteins which were identified in ovary during all five stages of post-engorgement
The results of the KEGG pathway analysis on the proteins of the four Clusters with different trends are shown in Supplementary Fig. 4. 21 proteins in Cluster 1 were involved in the ribosome pathway. During ovarian development, a large amount of vitellin is consumed and synthesized, while the demand for most other proteins is relatively reduced (Chinzei et al. 1983), causing the new proteins generated by the ovary through ribosomes to also be reduced, which is the main reason for the decreased expression levels of these proteins in the ribosome pathway. 6 proteins were involved in the necroptosis pathway. Necroptosis during development of an organism is a common cell death controlled by genes, representing a suicide protection mechanism (Frank and Vince 2019). It is as important as cell proliferation and plays a very important role in the growth and development of multicellular organisms. These proteins involved in the necroptosis pathway enable the organism to maintain homeostasis (O'Donnell et al. 2018), and play an important role in ovarian development.
In Cluster 2 and Cluster 4, serine/threonine-protein phosphatase PP1-beta catalytic subunit, Ras family protein, eIF2B-gamma protein, DNA repair protein xp-E, and other proteins were involved in the development of various diseases, such as pathogenic escherichia coli infection, herpes simplex virus infection, human immunodeficiency virus infection, viral carcinogenesis, proteoglycans in cancer, Epstein-Barr virus infection, hepatitis B.
In Cluster 3, 64 proteins were involved in various metabolic pathways, and their expression levels kept increasing with ovarian development. For example, in glycometabolism, the increases in transketolase, glyceraldehyde 3-phosphate dehydrogenase, and hexokinase can provide more energy for the organism. 13 proteins were involved in the lysosome pathway. A lysosome is an organelle that decomposes biological macromolecules, which contains various hydrolases, such as aspartic protease, cysteine protease, serine protease, and carboxypeptidase. These proteins are capable of decomposing Vn to provide energy for embryo development, thus playing an important role during ovarian development.
We also performed the KEGG pathway analysis on the 545 phosphorylated proteins corresponding to the 1125 differentially expressed phosphorylated peptides (Table 1). The involved pathways included the mTOR signalling pathway and PI3K-AKT signalling pathway, which play important roles in cell growth and regulation; the Hippo signalling pathway, which regulates organ size by regulating cell proliferation, apoptosis, and stem cell self-renewal capacity; pathways related to cell growth and development, such as progesterone-mediated oocyte maturation; ubiquitin-mediated proteolysis pathway; endocytosis and autophagy pathway, which can remove the apoptotic or necrotic cells.
Discussion
Female tick increases in size during blood feeding and expands to more than 100 times compared with its original size during the rapid feeding phase (Reuben Kaufman 2007). After engorgement, the tick ovary enters the rapid development stage (Ullah and Kaufman 2014; Yang et al. 2014). To investigate the physiological regulation mechanism of the rapid developmental stage of the tick ovary, we systematically investigated the dynamic changes in expression levels of total proteins and those of protein phosphorylation during the rapid ovarian development of engorged female ticks using DIA quantitative proteomic technology. The results of quantitative proteomics (Supplementary Table 1) showed that with the continuous development of the ovary of female ticks after engorgement, the expression of a large number of proteins in the ovary began to down-regulated. The expression of a few proteins such as Vn was gradually up-regulated.
GO annotation analysis of the identified differentially expressed proteins in the ovaries during different stages of post-engorgement were most belonged to nucleic acid binding, hydrolase, enzyme modulator, transferase, and so on. KEGG signaling pathway analysis revealed that transketolase, glyceraldehyde 3-phosphate dehydrogenase, and hexokinase were involved in metabolism pathways, providing energy for ovary development. Other proteins belong to Lysosomal hydrolytic enzymes, such as aspartic protease, cysteine protease, and serine carboxypeptidase, were involved in the degradation of Vn protein during the subsequent development of eggs. In addition, the differentially expressed phosphorylated proteins such as protein kinase C, RNA-binding protein, translation initiation factor 4B, serine/threonine protein kinase 3 mainly involved in the mTOR, PI3K-AKT, and Hippo signaling pathways, which involved in cell growth and development. In addition, some phosphorylated proteins such as cytoplasmic polyadenylation element binding protein and anaphase-promoting complex subunit take part in progesterone-mediated oocyte maturation.
Vitellogenesis is an important part of tick reproductive process and directly affects its reproductive capacity. Vn is a heme glycolipid complex protein, which is the major reserve protein present in yolk granules, constitutes the major yolk protein (Estrela et al. 2010). Vn is a nutrient source for embryo development, survival, and reproduction of ticks, which is very important for the development of tick embryos (Mitchell et al. 2019). The metabolites produced by the degradation of Vn even provide nutrients for the newly hatched larvae (Seixas et al. 2003). Therefore, during ovarian development of the engorged H. longicornis, a large amount of Vn is present. Vg as the precursor of Vn is usually synthesized at extra-ovarially (Khalil et al. 2011). The formation and ingestion of Vg are the key links in vitellogenesis. Vg is abundantly present in the body of tick, is mainly synthesized in the fat body and midgut of the mated female tick, and rarely in the ovary (Taylor et al. 1991; Thompson et al. 2007). After Vg is released to the haemolymph (Coons et al. 1989), a surface receptor called the Vg receptor captures and endocytose them into the oocytes (Mitchell et al. 2019). Through processing and post-translational modification, Vg is converted to Vn (James and Oliver 1997). In ticks, ecdysteroids activate vitellogenesis and regulate female ticks reproduction by regulating the expression of Vg gene and regulating the synthesis and release of Vg protein into haemolymph (Cabrera et al. 2009). After that, a series of regulatory processes of vitellogenesis are carried out, which requires the coordination of various proteins, so as to achieve the rapid and efficient formation of eggs.
Vn is hydrolyzed into raw materials such as amino acids needed by embryo development under the action of various hydrolytic enzymes in ovaries (Giorgi et al. 1999). In insect, many hydrolytic enzymes related to Vn degradation, such as aspartic protease, cysteine protease, serine protease, and carboxypeptidase, have been extensively investigated (Guo et al. 2019). Most proteases hydrolysing Vn in oocytesare present in an inactive form, these proteases are preserved as inactive zymogens (Takahashi et al. 1993), and converted to the active form through the acidification of yolk granules (Fagotto 1991, 1995). Compared with insects, there are relatively few studies on the digestion of Vn in ticks. It was found that aspartic endopeptidases and cysteine endopeptidase play an important role in the digestion of Vn in Rhipicephalus microplus (Estrela et al. 2010; Seixas et al. 2003; Nascimento-Silva et al. 2008). In this study, we found that in the ovarian development of the engorged H. longicornis, the expression levels of aspartic protease, cysteine protease, and serine carboxypeptidase gradually increased with ovary development, suggesting that these enzymes are continuously accumulated during the rapid ovary development for the subsequent Vn decomposition.
Proteins related to aspartic protease that were identified during ovarian development, which belongs to the lysosomal enzyme group that has been shown to be involved in the Vn hydrolysis process of tetrapods (Opresko and Karpf 1987), and the Vn hydrolysis process of chicken oocytes (Retzek et al. 1992). It was showed that aspartic protease plays a major role in Vn decomposition, whose activity is controlled by the acidification of yolk platelets (Abreu et al. 2004). In this study, we found that during ovarian development, the expression of aspartic protease kept increasing, and the expression at 48 h post-engorgement was 53.2 times of that at 0 h post-engorgement, indicating that during ovarian development, a large amount of aspartic protease is accumulated, which lays the foundation for the subsequent Vn decomposition during embryo development.
Cysteine proteases are widely present in lysosomes of organisms (Boulangé et al. 2001), they are involved in phagocytosis and scavenge excess cellular materials, thereby playing an important role in degradation and processing processes of protein (Castro-Gomes et al. 2016). Cysteine proteases have been found in various parasitic species and are involved in many physiological processes, such as protein hydrolysis, autophagy, and escape and regulation of the host immune response (Gomes et al. 2017; Siqueira-Neto et al. 2018). They also play a very important role in insect metamorphosis, especially during embryogenesis and tissue remodelling stages, while they are also involved in the circulation of nutrients (Sojka et al. 2011). In this study, we found that the expression of cysteine protease increased with the ovarian development, and the expression at 48 h post-engorgement was 2.6 times of that at 0 h post-engorgement, which may play an important role in embryonic development by degrading Vn.
Carboxypeptidases are important digestive enzymes in insects and are peptide exonucleases that degrade peptide chains from the C-terminus and thus release free amino acids (Zhang et al. 2019). According to the catalytic centre, carboxypeptidases can be divided into three major categories: serine carboxypeptidase (SCP), cysteine carboxypeptidase, and metal carboxypeptidase. SCP, also known as acidic carboxypeptidase, is a proteolytic enzyme in eukaryotes; under acidic conditions, SCP has terminal proteolytic enzyme activity and is involved in protein processing, modification, and degradation (Breddam 1986). Thus far, only a few SCPs have been studied in insects and were found to be involved in protein digestion in the gut of animals (Bown et al. 1998) and in Vn degradation in mosquito oocytes (Cho et al. 1991). SCPs play an important role in the digestion of the host's blood-meal and also take part in the proteolytic cascades for hemoglobin degradation in H. longicornis (Motobu et al. 2007). In ovarian development of the engorged H. longicornis, the expression levels of SCPs kept increasing, and that of SCP at 48 h post-engorgement was 7.6 times that at 0 h post-engorgement, indicating that a large quantity of this SCP accumulated during ovarian development and may participated in Vn degradation after embryonic development was initiated.
The expression levels of proteins in Cluster 1 assumed a sustained down-regulated trend during ovarian development. The synthesis and accumulation of a large quantity of Vn during ovarian maturation provided nutrients for the subsequent embryonic development, while the gradually decreased expression levels of a large number of other proteins showed that ovary can mobilize more energy and raw materials for Vn synthesis (Chinzei et al. 1983). The expression levels of many ribosome-related proteins are down-regulated. Ribosomal-related proteins, referring to all proteins involved in the formation of a ribosome, are major components of the ribosome and play important roles in the biosynthesis of proteins. In the late stage of ovarian development, the protein types in the ovary were decreased, and the needed ribosomes also decreased accordingly, so ribosome-related proteins were down-regulated. Moreover, the expression levels of a large number of eukaryotic translation initiation factors were also down-regulated. The protein translation initiation process of eukaryotes is a complex process in which many proteins participate, collectively referred to as translation initiation factors. Because the number of protein type required in the late stage of ovarian development was reduced, the expression levels of eukaryotic translation initiation factors also exhibited a down-regulated trend.
During ovarian development, the phosphorylation levels of the pT222 and pT226 sites of the heat shock protein 70 kDa protein (Hsp70) kept increasing. Hsp70 is a conserved and ubiquitous member of the heat shock protein family and plays an essential role in maintaining cell viability (Morano 2007) by protecting cells from heat or oxidative stress and directly inhibiting apoptosis (Beere et al. 2000). Hsp70 is a molecular chaperone, and phosphorylated Hsp70 is involved in the folding of proteins during signal transduction while guiding the ubiquitin-mediated degradation of unfolded proteins (Muller et al. 2013), and playing an important regulatory role in the normal progression of the cell cycle. Therefore, we believe that this protein plays a key role in ovarian development.
During ovarian development, the phosphorylation level of the pT420 site of the engulfment and cell motility protein (ELMO) was increasing. The phosphorylated ELMO protein promotes cell migration (Grimsley et al. 2004). The members of ELMO family are present in fungus, nematoda, arthropoda, and higher vertebrates (Brzostowski et al. 2009). ELMO, characterized by an atypical C-terminal PH domain and a proline-rich motif that are associated with ELMO binding to the dedicator of cytokinesis (DOCK) protein (Patel et al. 2011), is a key protein in the chemokine signalling pathway. The phosphorylated ELMO protein binds to the DOCK2 protein to form a complex, thereby activating Rac proteins, which in turn regulate cell division and cell migration via actin.
With the development of the ovary, the expression levels of a large number of phosphorylated Vg and Vg precursor related proteins are up-regulated, and these proteins expression can be up-regulated more than 100 times at 48 h post-engorgement. An increase in these proteins will contribute to the development of Vn and promote ovarian development.
Protein expression changes provide various assembly and control mechanisms for the development of the body, while protein phosphorylation provides a unique, fast, reversible, energy-efficient mechanism to control the process of body development (Hawkins et al. 2019). Phosphorylation of proteins increases the diversity of proteins, allowing one protein to perform multiple functions and thus reduce the consumption of the resources in the body by not producing a large number of proteins. During the rapid development of the ovary, a large amount of energy and raw materials need to be accumulated, while proteins involved in carbohydrate metabolism, lipid metabolism, cholesterol metabolism, purine base metabolism, cytoskeleton synthesis, and signal transduction pathways need to work coordinately. The efficiency of the body is significantly improved by the phosphorylation of proteins. The investigation of the phosphorylation characteristics of these proteins is of great importance for better understanding the molecular mechanisms that regulate the reproductive development of ticks.
Conclusion
In this study, DIA quantitative proteomics method was used to study the changes of protein expression and phosphorylation during the ovarian development of female H. longicornis. This will provide a deeper understanding of the synergistic patterns of proteins in the whole process of ovarian development. The research results may provide a new protein target for the control of ticks. It could even have profound implications for the prevention of tick-borne diseases.
References
Abreu LA, Valle D, Manso PP, Façanha AR, Pelajo-Machado M, Masuda H, Vaz I Jr, Lenzi H, Oliveira PL, Logullo C (2004) Proteolytic activity of Boophilus microplus yolk pro-Cathepsin D (BYC) is coincident with cortical acidification during embryogenesis. Insect Biochem Mol Biol 34:443–449
Bartíková P, Holíková V, Kazimírová M, Štibrániová I (2017) Tick-borne viruses. Acta Virol 61:413–427
Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2:469–475
Boulangé A, Serveau C, Brillard M, Minet C, Gauthier F, Diallo A, Lalmanach G, Authié E (2001) Functional expression of the catalytic domains of two cysteine proteinases from Trypanosoma congolense. Int J Parasit 31:1435–1440
Bown DP, Wilkinson HS, Gatehouse JA (1998) Midgut carboxypeptidase from Helicoverpa armigera (Lepidoptera: Noctuidae) larvae: enzyme characterization, cDNA cloning and expression. Insect Biochem Mol Biol 28:739–749
Breddam K (1986) Serine carboxypeptidases. A review. Carlsberg Res Commun 51:83
Brzostowski JA, Fey P, Yan J, Isik N, Jin T (2009) The Elmo family forms an ancient group of actin-regulating proteins. Commun Integr Biol 2:337–340
Cabrera AR, Donohue KV, Roe RM (2009) Regulation of female reproduction in mites: a unifying model for the Acari. J Insect Physiol 55:1079–1090
Castro-Gomes T, Corrotte M, Tam C, Andrews NW (2016) Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS ONE 11:e0152583
Cayol C, Koskela E, Mappes T, Siukkola A, Kallio ER (2017) Temporal dynamics of the tick Ixodes ricinus in northern Europe: epidemiological implications. Parasit Vectors 10:166
Chinzei Y, Chino H, Takahashi K (1983) Purification and properties of vitellogenin and vitellin from a tick, Ornithodoros moubata. J Comp Physiol B 152:13–21
Cho WL, Deitsch KW, Raikhel AS (1991) An extraovarian protein accumulated in mosquito oocytes is a carboxypeptidase activated in embryos. Proc Natl Acad Sci USA 88:10821–10824
Coons LB, Lamoreaux WJ, Roselldavis R, Tarnowski BI (1989) Onset of vitellogenin production and vitellogenesis, and their relationship to changes in the midgut epithelium and oocytes in the tick Dermacentor variabilis. Exp Appl Acarol 6:291–305
de la Fuente J (2018) Controlling ticks and tick-borne diseases looking forward. Ticks Tick Borne Dis 9:1354–1357
Estrela AB, Seixas A, Teixeira Vde O, Pinto AF, Termignoni C (2010) Vitellin- and hemoglobin-digesting enzymes in Rhipicephalus (Boophilus) microplus larvae and females. Comp Biochem Physiol B 157:326–335
Fagotto F (1991) Yolk degradation in tick eggs-III: Developmentally regulated acidification of the yolk spheres. Dev Growth Differ 33:57–66
Fagotto F (1995) Regulation of yolk degradation, or how to make sleepy lysosomes. J Cell Sci 108:3645–3647
Frank D, Vince JE (2019) Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 26:99–114
Giorgi F, Bradley JT, Nordin JH (1999) Differential vitellin polypeptide processing in insect embryos. Micron 30:579–596
Gomes CB, Silva FS, Charret KD, Pereira BA, Finkelstein LC, Santos-de-Souza R, de Castro Côrtes LM, Pereira MC, de Oliveira R, Jr FO, Alves CR (2017) Increasing in cysteine proteinase B expression and enzymatic activity during in vitro differentiation of Leishmania (Viannia) braziliensis: first evidence of modulation during morphological transition. Biochimie 133:28–36
Grimsley CM, Kinchen JM, Tosello-Trampont AC, Brugnera E, Haney LB, Lu M, Chen Q, Klingele D, Hengartner MO, Ravichandran KS (2004) Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem 279:6087–6097
Guo W, Wu Z, Yang L, Cai Z, Zhao L, Zhou S (2019) Juvenile hormone-dependent Kazal-type serine protease inhibitor Greglin safeguards insect vitellogenesis and egg production. FASEB J 33:917–927
Hawkins LJ, Wang M, Zhang B, Xiao Q, Wang H, Storey KB (2019) Glucose and urea metabolic enzymes are differentially phosphorylated during freezing, anoxia, and dehydration exposures in a freeze tolerant frog. Comp Biochem Physiol Part D 30:1–13
James AM, Oliver JH Jr (1997) Purification and partial characterization of vitellin from the black-legged tick, Ixodes scapularis. Insect Biochem Mol Biol 27:639–649
Khalil SM, Donohue KV, Thompson DM, Jeffers LA, Ananthapadmanaban U, Sonenshine DE, Mitchell RD, Roe RM (2011) Full-length sequence, regulation and developmental studies of a second vitellogenin gene from the American dog tick, Dermacentor variabilis. J Insect Physiol 57:400–408
Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R (2018) Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol 14:e8126
Mitchell RD 3rd, Sonenshine DE, Pérez de León AA (2019) Vitellogenin receptor as a target for tick control: a mini-review. Front Physiol 10:618
Morano KA (2007) New tricks for an old dog: the evolving world of Hsp70. Ann N Y Acad Sci 1113:1–14
Motobu M, Tsuji N, Miyoshi T, Huang X, Islam MK, Alim MA, Fujisaki K (2007) Molecular characterization of a blood-induced serine carboxypeptidase from the ixodid tick Haemaphysalis longicornis. FEBS J 274:3299–3312
Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, Vojtesek B (2013) C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 32:3101–3110
Nascimento-Silva MC, Leal AT, Daffre S, Juliano L, da Silva VI, Jr P-S, Oliveira PL, Sorgine MH (2008) BYC, an atypical aspartic endopeptidase from Rhipicephalus (Boophilus) microplus eggs. Comp Biochem Physiol B 149:599–607
O'Donnell JA, Lehman J, Roderick JE, Martinez-Marin D, Zelic M, Doran C, Hermance N, Lyle S, Pasparakis M, Fitzgerald KA, Marshak-Rothstein A, Kelliher MA (2018) Correction: dendritic cell RIPK1 maintains immune homeostasis by preventing inflammation and autoimmunity. J Immunol 200:3020–3021
Opresko LK, Karpf RA (1987) Specific proteolysis regulates fusion between endocytic compartments in Xenopus oocytes. Cell 51:557–568
Otaki H, Sonobe J, Murphy M, Cavalleri D, Seewald W, Drake J, Nanchen S (2018) Laboratory evaluation of the efficacy of lotilaner (Credelio™) against Haemaphysalis longicornis infestations of dogs. Parasit Vectors 11:448
Patel M, Pelletier A, Côté JF (2011) Opening up on ELMO regulation: new insights into the control of Rac signaling by the DOCK180/ELMO complex. Small GTPases 2:268–275
Rachinsky A, Guerrero FD, Scoles GA (2007) Differential protein expression in ovaries of uninfected and Babesia-infected southern cattle ticks, Rhipicephalus (Boophilus) microplus. Insect Biochem Mol Biol 37:1291–1308
Raikhel AS, Dhadialla TS (1992) Accumulation of yolk proteins in insect oocytes. Annu Rev Entomol 37:217–251
Retzek H, Steyrer E, Sanders EJ, Nimpf J, Schneider WJ (1992) Molecular cloning and functional characterization of chicken Cathepsin D, a key enzyme for yolk formation. DNA Cell Biol 11:661–672
Reuben Kaufman W (2007) Gluttony and sex in female ixodid ticks: how do they compare to other blood-sucking arthropods? J Insect Physiol 53:264–273
Rigbolt KT, Vanselow JT, Blagoev B (2011) GProX, a user-friendly platform for bioinformatics analysis and visualization of quantitative proteomics data. Mol Cell Proteomics 10(8):O110–007450
Rodríguez PBR, Cruz RR, García DID, Gutiérrez RH, Quintanilla REL, Sahagún DO, Castillo CG, Ortega AG, Rodríguez SEH, Cardona AV, Velázquez MM (2016) Identification of immunogenic proteins from ovarian tissue and recognized in larval extracts of Rhipicephalus (Boophilus) microplus, through an immunoproteomic approach. Exp Parasitol 170:227–235
Rosenberger G, Bludau I, Schmitt U, Heusel M, Hunter CL, Liu Y, MacCoss MJ, MacLean BX, Nesvizhskii AI, Pedrioli PGA, Reiter L, Röst HL, Tate S, Ting YS, Collins BC, Aebersold R (2017) Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat Methods 14:921–927
Seixas A, Dos Santos PC, Velloso FF, Da Silva VI, Jr MA, Horn F, Termignoni C (2003) A Boophilus microplus vitellin-degrading cysteine endopeptidase. Parasitology 126:155–163
Siqueira-Neto JL, Debnath A, McCall LI, Bernatchez JA, Ndao M, Reed SL, Rosenthal PJ (2018) Cysteine proteases in protozoan parasites. PLoS Negl Trop Dis 12:e0006512
Sojka D, Francischetti IM, Calvo E, Kotsyfakis M (2011) Cysteine proteases from blood feeding arthropod ectoparasites. Adv Exp Med Biol 712:177–191
Takahashi SY, Yamamoto Y, Shionoya Y, Kageyama T (1993) Cysteine proteinase from the eggs of the silkmoth, Bombyx mori: identification of a latent enzyme and characterization of activation and proteolytic processing in vivo and in vitro. J Biochem 114:267–272
Taylor D, Chinzei Y, Miura K, Anado K (1991) Vitellogenin synthesis, processing and hormonal regulation in the tick, Ornithodoros parkeri (Acari: Argasidae). Insect Biochem 21:723–733
Thompson DM, Khalil SMS, Jeffers LA, Sonenshine DE, Mitchell RD, Osgood CJ, Michael Roe R (2007) Sequence and the developmental and tissue-specific regulation of the first complete vitellogenin messenger RNA from ticks responsible for heme sequestration. Insect Biochem Mol Biol 37:363–374
Ullah SA, Kaufman WR (2014) Salivary gland degeneration and ovarian development in the Rocky Mountain wood tick, Dermacentor andersoni Stiles (Acari: Ixodidae). I. Post-engorgement events. Ticks Tick Borne Dis 5:569–574
Wada T, Ishiwata K, Koseki H, Ishikura T, Ugajin T, Ohnuma N, Obata K, Ishikawa R, Yoshikawa S, Mukai K, Kawano Y, Minegishi Y, Yokozeki H, Watanabe N, Karasuyama H (2010) Selective ablation of basophils in mice reveals their nonredundant role in acquired immunity against ticks. J Clin Invest 120:2867–2875
Wang H, Zhang X, Wang X, Zhang B, Wang M, Yang X, Han X, Wang R, Ren S, Hu Y, Liu J (2018) Comprehensive analysis of the global protein changes that occur during salivary gland degeneration in female Ixodid ticks Haemaphysalis longicornis. Front Physiol 9:1943
Xavier MA, Tirloni L, Pinto AFM, Diedrich JK, Yates JR 3rd, Mulenga A, Logullo C, da Silva VI, Jr SA, Termignoni C (2018) A proteomic insight into vitellogenesis during tick ovary maturation. Sci Rep 8:4698
Yang XL, Yu ZJ, Gao ZH, Yang XH, Liu JZ (2014) Morphological characteristics and developmental changes of the ovary in the tick Haemaphysalis longicornis Neumann. Med Vet Entomol 28:217–221
Zhang X, Zhang L, Li J (2019) Peptide-modified nanochannel system for carboxypeptidase B activity detection. Anal Chim Acta 1057:36–43
Acknowledgements
The authors are very grateful to Dr Baowen Zhang for her technical support in the use of mass spectrometer and helping in data analyse. This work was supported by the Natural Science Foundation for Excellent Youth Scholars of Hebei Province of China (No. C2017205135), the Natural Science Fund for Distinguished Young Scholars of Hebei Normal University (No. L2017J04).
Author information
Authors and Affiliations
Contributions
HW designed experiments, analyzed the data, performed experiments, and wrote the initial manuscript. MW, YH, ML, XZ, performed experiments. XW, XX, QX prepared figures. JL designed experiments, correction of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Animal Ethics Committee of Hebei Normal University (Protocol Number: 165031) as complying with the animal protection law of the People’s Republic of China.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10493_2020_469_MOESM1_ESM.jpg
Supplementary Figure S1. GO “Protein Class” analysis of differentially expressed 506 proteins and 545 phosphorylated proteins which were identified in ovary during all five stages of post-engorgement. a GO “Protein Class” analysis of differentially expressed proteins. b GO “Protein Class” analysis of all differentially expressed phosphorylated proteins (JPEG 322 kb)
10493_2020_469_MOESM2_ESM.jpg
Supplementary Figure S2. GO functional annotations for the differentially expressed proteins in the ovaries of female H. longicornis at five stages of post-engorgement (JPEG 339 kb)
10493_2020_469_MOESM3_ESM.jpg
Supplementary Figure S3. GO functional annotations for the differentially expressed phosphorylated proteins in the ovaries of female H. longicornis during five stages of post-engorgement (JPEG 375 kb)
10493_2020_469_MOESM4_ESM.jpg
Supplementary Figure S4. KEGG pathway analysis of the differentially expressed proteins in 4 Clusters. a-d KEGG pathway analysis of proteins in Cluster 1 to Cluster 4 (JPEG 390 kb)
10493_2020_469_MOESM5_ESM.xlsx
Supplementary Table S1. Statistics for protein quantification information of the common proteins were identified at five stages. These proteins were considered highly reliable, which at least 2 unique peptides were identified and have a CV < 20% among four replicates in each group (XLSX 167 kb)
10493_2020_469_MOESM6_ESM.xlsx
Supplementary Table S2. The data of phosphorylated peptides corresponding to differentially expressed phosphorylated proteins at five stages of post-engorgement. These phosphorylated peptides were considered highly reliable, which were have a CV < 20% among four replicates in each group (XLSX 156 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, M., Hu, Y., Li, M. et al. A proteomics analysis of the ovarian development in females of Haemaphysalis longicornis. Exp Appl Acarol 80, 289–309 (2020). https://doi.org/10.1007/s10493-020-00469-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-020-00469-3