
Abstract
Haemaphysalis qinghaiensis as an endemic species in China mainly infests domestic animals and causes great harm to animals and humans in Northwestern plateau. However, there is no information about genetic diversity within the recently established populations of this tick species. Therefore, the present study analyzed the fragments of mitochondrial 16S rDNA, COI and the nuclear gene ITS1 of 56 H. qinghaiensis ticks across four regions of China which are main endemic areas of this species. Analysis showed 98.1–100% (16S rDNA), 97.9–100% (COI), 99.7–100% (ITS1) identity within individuals. For these sequences, 9, 15 and 8 haplotypes were found for 16S rDNA, COI and ITS1, respectively. Ticks from Yongjing were the most variable group, followed by Lintan, Huangyuan, and Tianzhu. Based on parallel analysis of the mitochondrial and nuclear genetic diversity of H. qinghaiensis, our results indicated that mitochondrial markers (especially COI) were much more useful than nuclear ITS for intraspecific genetic variability analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ticks are serious vectors of numerous human and animal pathogens, which are currently considered to be second only to mosquitoes. Associating tick haplotypes with its borne pathogens and the environment is very useful to control ticks and tick-borne disease. It is known Haemaphysalis is the second largest tick genus of the world, and the largest genus in China (Chen et al. 2010), many species of which closely related to humans. However, seldom studies of population genetic variation for Haemaphysalis tick species have been carried out. Haemaphysalis qinghaiensis Teng is a Chinese endemic species, and widely distributed in the western plateau (Teng and Jiang 1991; Chen et al. 2010, 2014a, b). This species is a three host tick, and often infest medium-large-sized domestic animals including sheep, goats, cattle, yaks, horses, mule and donkey, and sometimes humans. It was reported that H. qinghaiensis is an important vector of Theileria and Babesia, causing great harm to the animal husbandry and the health of animals and humans in Northwestern China (Guan et al. 2002, 2010; Li et al. 2007, 2009; Niu et al. 2017).
16S rDNA is frequently used to analyze the genetic variation within and among tick populations (Krakowetz et al. 2010, 2011; Dantastorres et al. 2013; Low et al. 2015; Arayaanchetta et al. 2015). Other genes such as 12S rDNA, COI, COIII, cytb, 18S rDNA, ITS1 and ITS2 are also used in genetic diversity studies for some tick species (Crosbie et al. 1998; Kempf et al. 2009; Tian et al. 2011; Gomezdiaz et al. 2012; Livanova et al. 2015). The aim of the present study was to determine the genetic polymorphisms of H. qinghaiensis from various ecoregions of China by parallel analyzing parts of COI, 16S rDNA, and ITS1 gene sequences.
Materials and methods
Tick collection
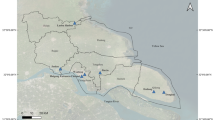
Huangyuan, Lintan, Yongjing and Tianzhu are main endemic areas of H. qinghaiensis in China. Their ecology and environment are similar to each other (continental climate and drought), but with different altitude. The percentage altitude of Tianzhu, Huangyuan, Lintan and Yongjing was 3500, 3100, 2825, and 1600 m, respectively. Therefore, H. qinghaiensis ticks were collected and sampled from these distinct locations with about 70,000 km2 sampling area (Fig. 1, Table 1). Only ticks from Lintan were collected on vegetation by flagging, while others were captured from cattle. These specimens were identified by morphological characteristics (Chen et al. 2014a, b).

Sampling sites of Haemaphysalis qinghaiensis ticks
DNA isolation and amplification
The genomic DNAs of collected specimens (Table 1) were extracted using the DNeasy tissue kit (Qiagen, Hilden, Germany). The primer sets of 16S rDNA, COI and ITS1 gene sequences were synthesized by Shanghai Sangon Biotechnology (Shanghai, China). Briefly, the primers 16SF (5′-CTGCTCAATGATTTTTTAAATTGCTGTGG-3′) and the reverse primer 16SR (5′-CCGGTCTGAACTCAGATCAAGT-3′) (Black and Piesman 1994) were used for 16S rDNA amplification, primers COIF1 (5′-AATTTACAGTTTATCGCCT-3′) and COIR (5′-CATACAATAAAGCCTAATA-3′) (Black and Piesman 1994) were used for COI gene amplification, and primers ITS1F (5′-TCATAAGCTCGCGTTGATT-3′) and ITS1R (5′-AGCTGGCTGCGTTCTTCAT-3′) (Chitimia et al. 2009) were used for ITS1 gene amplification. The PCR conditions were already described by Chen et al. (2012), and their products were about 450, 880 and 1400 bp for 16S rDNA, COI and ITS1, respectively. Negative controls (no template) were run in all PCR experiments. QIAGEN purification columns were used to purify 100 μl of PCR product according to the manufacturer’s instructions. Both strands of the purified PCR product were sequenced twice by Shanghai Sangon Biotechnology.
Data analysis
Using the software BioEdit 5.09 (Hall 1999) edited the sequencing results and verified the nucleotide polymorphisms. The determined sequences were aligned with the CLUSTALX 2.0.11 software. The distribution of genetic variation between groups was analyzed by the Kimura two-parameter model, as implemented in MEGA 6 (Tamura et al. 2013). Haplotypes were defined by TCS 1.13 when they differed by at least one base pair including indels (Clement et al. 2000). Genealogical relationships among haplotypes of H. qinghaiensis were constructed using TCS 1.13 (Clement et al. 2000).
Results
In total, 56 adult ticks of H. qinghaiensis were collected from four distinct locations, namely Huangyuan (20), Lintan (23), Yongjing (6) and Tianzhu (7). In order to compare the genetic diversity of mitochondrial 16S rDNA and COI, and the nuclear gene ITS1 in parallel, each individual tick was amplified all of the three gene fragments, including 16S rDNA (54 sequences), COI (42 sequences) and ITS1 (45 sequences) (Table 1). The alignment of 16S rDNA, COI and ITS1 sequences resulted in a total of 444, 783 and 1386 data matrix (including gaps), respectively. The summary statistics for the three gene fragments are described in Table 2.
Analysis of mt 16S rDNA, COI, and the nuclear gene ITS1 sequences showed 98.1–100% (16S rDNA), 97.9–100% (COI), 99.7–100% (ITS1) identity within H. qinghaiensis individuals. For these sequences, 9, 15 and 8 haplotypes were found for 16S rDNA, COI and ITS1, respectively (Figs. 2, 3, 4). Using the Kimura two-parameter substitution analysis, the value of intra-specific genetic distances was 0–0.011, 0–0.02, 0–0.004 for 16S rDNA, COI and ITS1, respectively.

Genealogical relationships of Haemaphysalis qinghaiensis based on nucleotide variability sites of 16S rDNA using TCS (Clement et al. 2000)

Genealogical relationships of Haemaphysalis qinghaiensis based on nucleotide variability sites of COI using TCS (Clement et al. 2000)

Genealogical relationships of Haemaphysalis qinghaiensis based on nucleotide variability sites of ITS1 using TCS (Clement et al. 2000)
As little intraspecific differences of ITS1 between H. qinghaiensis ticks, a comparing species of H. flava (KJ156375) were also used for the analysis of sequence identity and inter-specific genetic distances in this study. The identity of ITS1 sequences between H. flava and H. qinghaiensis ticks was 97.7–97.9%, and their interspecific distance was 0.01–0.02. This interspecific difference of ITS1 sequences between H. flava and H. qinghaiensis is similar to intraspecific difference of 16S rDNA and COI sequences among H. qinghaiensis ticks, although the interspecific difference is larger than intraspecific difference for ITS1 analysis.
Of the 444 characters used for 16S rDNA analysis, 430 characters were constant and 14 were mutations (Tables 2, 3). The reconstruction of the network showed 9 detected 16S rDNA haplotypes (Fig. 2). The TCS spanning tree rooted H. qinghaiensis haplotype HQS1 as the ancestral haplotype. The two haplotypes (HQS1 and HQS2) combined 85% of all sequences which were found in most sampling sites. Additionally, HQS1 was found in all studied locations, whereas HQS4–HQS9 was unique to the sampling sites. The largest number of mutation sites was 5, and the Haplotypes were HQS4 (LT48) and HQS7 (YJ3) (Table 3). Haemaphysalis qinghaiensis from Yongjing (6 ticks) was the highest various group with 5 haplotypes, followed by that from Lintan (5 haplotypes in 21 ticks), Huangyuan (3 haplotypes in 20 ticks) and Tianzhu (1 haplotype in 7 ticks).
The aligned 42 COI derived gene sequences resulted in a 783 bp data matrix. Of these, 749 characters were constant and 32 were mutations (Tables 2, 4). The reconstruction of the network showed 15 detected COI haplotypes (Fig. 3). The TCS spanning tree rooted H. qinghaiensis haplotype HQC1 as the ancestral haplotype. There was no haplotype found in all studied locations, and HQC4–HQC15 was unique to the sampling sites. The two haplotypes of HQC1and HQC2 combined 64% of all sequences which were found in 3 sampling sites (Huangyuan, Lintan and Tianzhu), and 2 sampling sites (Lintan and Tianzhu), respectively. Additionally, H. qinghaiensis from Yongjing (5 ticks) was the highest various group with 5 haplotypes, and every of these haplotypes was unique. The largest number of mutation sites was 11 with the Haplotype HQC9 (LT50), and followed 9 with the haplotype HQC2 (LT1, LT5, LT6, LT9, LT10, LT12, LT19, LT28, LT41, LT46, TZ4, TZ5, TZ7) and HQC12 (YJ3) (Table 4).
The lengths of the aligned sequences of ITS1 were measured 1386 characters. Tables 2 and 5 show 1376 characters are constant and 10 are mutations. The reconstruction of the network showed 8 detected ITS1 haplotypes (Fig. 4). The TCS spanning tree rooted H. qinghaiensis haplotype HQI1 as the ancestral haplotype. The two haplotypes (HQI1 and HQI2) combined 87% of all sequences which were found in most sampling sites. Additionally, HQI1 was found in all studied locations, whereas HQI3–HQI8 was unique to the sampling sites. The largest number of mutation sites was 3 with the Haplotype HQI8 (YJ4), while each of most others was only one. In total, there were 2 haplotypes in 6 ticks from Yongjing, 1 haplotype in 7 ticks from Tianzhu, 3 haplotypes in 14 ticks from Huangyuan and 7 haplotypes in 18 ticks from Lintan.
The analysis of haplotypes of the three genes showed that H. qinghaiensis from Yongjing is the most genetically diverse population, and followed by Lintan, Huangyuan and Tianzhu (Table 6). Analysis of 16S rDNA, COI and ITS1 sequences showed 98.6–100% (98.5–99.7%, 99.7–100%), 98.6–100% (98.2–100%, 99.7–100%), 99.5–100% (99.7–100%, 99.8–100%) and 100% (98.7–100%, 100%) identity within H. qinghaiensis individuals from Yongjing, Lintan, Huangyuan and Tianzhu, respectively. Meanwhile, analysis of mt 16S rDNA, COI, and the nuclear gene ITS1 sequences showed 98.1–100% (16S rDNA), 97.9–100% (COI), 99.7–100% (ITS1) identity within H. qinghaiensis individuals across the four regions. The genetic distance of 16S rDNA and ITS1 within and between the four regions was 0. Each genetic distance of COI within Yongjing, Lintan and Tianzhu was 0.01, while that within Huangyuan was 0. The genetic distance of COI between Yongjing and Huangyuan was 0, while others were 0.01.
Discussion
Up to now, seldom studies of genetic diversity for Haemaphysalis tick species have been carried out. H. qinghaiensis is an endemic species in western plateau of China and often infest domestic animals (Teng and Jiang 1991; Chen et al. 2010, 2014a, b). This study was first to analyze the genetic diversity of mitochondrial 16S rDNA, COI and nuclear ITS1 fragments of H. qinghaiensis ticks in parallel. Haemaphysalis qinghaiensis ticks from different regions are hard to be identified from one population to another by morphological and molecular features. However, the genetic polymorphism of H. qinghaiensis from the four regions is different from each other.
In conclusion, most ticks were divided into the first two haplotypes of 16S rDNA, COI and ITS1, and the percentage was 85, 64 and 87%, respectively. The large percentage of singleton haplotypes (6/9 for 16S rDNA, 12/15 for COI, 6/8 for ITS1), and consistency of singleton and multiple haplotypes across the ecoregions indicated that these H. qinghaiensis populations were intermixing among the sampling areas (Figs. 2, 3, 4). This was also reported in other tick species (Trout et al. 2009, 2010; Kulakova et al. 2014). In this study, the percentage altitude of sampling area namely Tianzhu, Huangyuan, Lintan and Yongjing was 3500, 3100, 2825, and 1600 m, respectively. Interestingly, our study identified H. qinghaiensis ticks from Yongjing as the most genetically diverse population, and followed by Lintan, Huangyuan and Tianzhu. The region of Yongjing had 5 haplotypes for both of 16S rDNA and COI, of which 4 and 5 were unique, respectively. The sequences of most ticks from Tianzhu were identical to each other and to ancestral haplotype, especially for 16S rDNA and COI sequences. The reason of the different genetic polymorphism of H. qinghaiensis may be due to the height of altitude. Comparing to ticks at high altitude, the activity of ticks is more frequently at low altitude for H. qinghaiensis. Additionally, Yongjing located convenient with frequent livestock mobility, which might cause the frequent exchange of genes for ticks. Meanwhile Tianzhu is a much more out-of-the-way and backward place. Additionally, there was no relationship between genetic diversity and sex.
The 16S rDNA, COI and ITS1 sequences of H. qinghaiensis individuals revealed 98.1–100, 97.9–100, 99.7–100% identity to each other, respectively. Similar results were reported for other tick species: 99–100% (16S rDNA) for H. longicornis (Chen et al. 2015), 99.3–100% (16S rDNA and COI) for Dermacentor everestianus (Chen et al. 2014a, b). The intraspecific distance between H. qinghaiensis ticks was 0–0.011 (16S rDNA), 0–0.02 (COI), 0–0.004 (ITS1), which was comparable with results of 16S rDNA obtained for H. concinna (0.001), H. longicornis (0–0.001) and H. punctata (0.007) (Zhang and Zhang 2014; Chen et al. 2015). Crosbie et al. (1998) evaluated the intraspecific variation in 16S rDNA of 11 specimens of D. hunter and found that except for 4 ticks differed by a single base other 7 ticks were identical to each other. Similar to this study of H. qinghaiensis, intraspecific variation in COI of H. longicornis was 0–0.027, which was also further than that of 16S rDNA (Lu et al. 2013).
Analysis of 16S rDNA, COI and ITS1 sequences showed 98.6–100% (98.5–99.7%, 99.7–100%), 98.6–100% (98.2–100%, 99.7–100%), 99.5–100% (99.7–100%, 99.8–100%) and 100% (98.7–100%, 100%) identity within H. qinghaiensis individuals from Yongjing, Lintan, Huangyuan and Tianzhu, respectively. Meanwhile, analysis of mt 16S rDNA, COI, and the nuclear gene ITS1 sequences showed 98.1–100, 97.9–100, 99.7–100% identity within H. qinghaiensis individuals across the regions, respectively. The genetic distance of 16S rDNA and ITS1 within and between the four regions was 0. Each genetic distance of COI within Yongjing, Lintan and Tianzhu was 0.01, while that within Huangyuan was 0. From the analysis above, the differences within each region is not larger than those across the regions.
In this study, an aligned 1386-bp segment of nuclear gene ITS1 used for analysis with the results of 10 mutation sites and 8 haplotypes, while an aligned much smaller segment of 16S rDNA (444) and COI (783) used for analysis with the results of more mutation sites (14 and 32) and haplotypes (9 and 15). The intraspecific variation of ITS1 was also much more conserved (0–0.004) than that of 16S rDNA (0–0.011) and COI (0–0.02). Beati et al. (2012) analyzed the genetic variability among Amblyomma variegatum ticks, and found that the ITS2 fragment consisted of only uninformative single nucleotide mutations. Similar results were also found by (Kulakova et al. 2014). They evaluated the intraspecific variation in 16S rDNA and ITS2 of 11 specimens of D. nuttalli and found that the genetic distance was 0.007 and 0.002, respectively. In conclusion, based on our parallel analysis of the mitochondrial and nuclear genetic diversity of H. qinghaiensis and reported studies of other tick species, mitochondrial markers (especially COI) should be much more useful than nuclear ITS1 and ITS2 for intraspecific genetic variability analysis.
References
Arayaanchetta A, Busch JD, Scoles GA, Wagner DM (2015) Thirty years of tick population genetics: a comprehensive review. Infect Genet Evol 29:164–179
Beati L, Patel J, Lucaswilliams H, Adakal H, Kanduma EG, Tembomwase E, Krecek R, Mertins JW, Alfred JT, Kelly S (2012) Phylogeography and demographic history of Amblyomma variegatum (Fabricius) (Acari: Ixodidae), the tropical bont tick. Vector Borne Zoonot 12:514–525
Chen Z, Yang X, Bu F, Yang X, Yang X, Liu J (2010) Ticks (Acari: Ixodoidea: Argasidae, Ixodidae) of China. Exp Appl Acarol 51:393–404
Chen Z, Yang X, Bu F, Liu J (2012) Morphological, biological and molecular characteristics of bisexual and parthenogenetic Haemaphysalis longicornis. Vet Parasitol 189:344–352
Chen Z, Li Y, Liu Z, Ren Q, Ma M, Luo J, Yin H (2014a) Scanning electron microscopy of all parasitic stages of Haemaphysalis qinghaiensis Teng, 1980 (Acari: Ixodidae). Parasitol Res 113:2095–2102
Chen Z, Li Y, Ren Q, Luo J, Liu Z, Zhou X, Liu G, Luo J, Yin H (2014b) Dermacentor everestianus Hirst, 1926 (Acari: Ixodidae): phylogenetic status inferred from molecular characteristics. Parasitol Res 113:3773–3779
Chen Z, Li Y, Ren Q, Liu Z, Luo J, Li K, Guan G, Yang J, Han X, Liu G (2015) Does Haemaphysalis bispinosa (Acari: Ixodidae) really occur in China? Exp Appl Acarol 65:249–257
Chitimia L, Lin R, Cosoroaba I, Braila P, Song HQ, Zhu XQ (2009) Molecular characterization of hard ticks from Romania by sequences of the internal transcribed spacers of ribosomal DNA. Parasitol Res 105:907–911
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Crosbie PR, Boyce WM, Rodwell TC (1998) DNA sequence variation in Dermacentor hunteri and estimated phylogenies of Dermacentor spp. (Acari: Ixodidae) in the New world. J Med Entomol 35:277–288
Dantastorres F, Latrofa MS, Annoscia G, Giannelli A, Parisi A, Otranto D (2013) Morphological and genetic diversity of Rhipicephalus sanguineus sensu lato from the New and Old Worlds. Parasite Vector 6:213
Gomezdiaz E, Morrispocock JA, Gonzalezsolis J, Mccoy KD (2012) Trans-oceanic host dispersal explains high seabird tick diversity on Cape Verde islands. Biol Lett 8:616–619
Guan G, Yin H, Luo J, Lu W, Zhang Q, Gao Y, Lu B (2002) Transmission of Babesia sp. to sheep with field-collected Haemaphysalis qinghaiensis. Parasitol Res 88:22–24
Guan G, Moreau E, Liu J, Hao X, Ma M, Luo J, Chauvin A, Hong Y (2010) Babesia sp. BQ1 (Lintan): molecular evidence of experimental transmission to sheep by Haemaphysalis qinghaiensis and Haemaphysalis longicornis. Parasitol Int 59:265–267
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Kempf F, Boulinier T, De Meeus T, Arnathau C, Mccoy KD (2009) Recent evolution of host-associated divergence in the seabird tick Ixodes uriae. Mol Ecol 18:4450–4462
Krakowetz CN, Dergousoff SJ, Chilton NB (2010) Genetic variation in the mitochondrial 16S rRNA gene of the American dog tick, Dermacentor variabilis (Acari: Ixodidae). J Vector Ecol 35:163–173
Krakowetz CN, Lindsay LR, Chilton NB (2011) Genetic diversity in Ixodes scapularis (Acari: Ixodidae) from six established populations in Canada. Ticks Tick Borne Dis 2:143–150
Kulakova NV, Khasnatinov MA, Sidorova EA, Adel Shin RV, Belikov SI (2014) Molecular identification and phylogeny of Dermacentor nuttalli (Acari: Ixodidae). Parasitol Res 113:1787–1793
Li Y, Luo J, Liu Z, Guan G, Gao J, Ma M, Dang Z, Liu A, Ren Q, Lu B (2007) Experimental transmission of Theileria sp. (China 1) infective for small ruminants by Haemaphysalis longicornis and Haemaphysalis qinghaiensis. Parasitol Res 101:533–538
Li Y, Luo J, Guan G, Ma M, Liu A, Liu J, Ren Q, Niu Q, Lu B, Gao J (2009) Experimental transmission of Theileria uilenbergi infective for small ruminants by Haemaphysalis longicornis and Haemaphysalis qinghaiensis. Parasitol Res 104:1227–1231
Livanova NN, Tikunov AY, Kurilshikov AM, Livanov SG, Fomenko NV, Taranenko DE, Kvashnina AE, Tikunova NV (2015) Genetic diversity of Ixodes pavlovskyi and I. persulcatus (Acari: Ixodidae) from the sympatric zone in the south of Western Siberia and Kazakhstan. Exp Appl Acarol 67:441–456
Low VL, Tay ST, Kho KL, Koh FX, Tan TK, Lim YAL, Ong BL, Panchadcharam C, Normarashid Y, Sofianazirun M (2015) Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasite Vector 8:341
Lu X, Lin X, Wang J, Qin X, Tian J, Guo W, Fan F, Shao R, Xu J, Zhang Y (2013) Molecular survey of hard ticks in endemic areas of tick-borne diseases in China. Ticks Tick Borne Dis 4:288–296
Niu Q, Liu Z, Yang J, Gao S, Pan Y, Guan G, Luo J, Yin H (2017) Genetic characterization and molecular survey of Babesia sp. Xinjiang infection in small ruminants and ixodid ticks in China. Infect Genet Evol 49:330–335
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Teng KF, Jiang ZJ (1991) Economic insect fauna of China. Fasc 39 Acari: Ixodidae. Science Press, Beijing (in Chinese)
Tian Z, Liu G, Xie J, Yin H, Luo J, Zhang L, Zhang P, Luo J (2011) Discrimination between Haemaphysalis longicornis and H. qinghaiensis based on the partial 16S rDNA and the second internal transcribed spacer (ITS-2). Exp Appl Acarol 54:165–172
Trout RT, Steelman CD, Szalanski AL (2009) Population genetics and phylogeography of Ixodes scapularis from Canines and Deer in Arkansas. Southwest Entomol 34:273–287
Trout RT, Steelman CD, Szalanski AL (2010) Population genetics of Amblyomma americanum (Acari; Ixodidae) collected from Arkansas. J Med Entomol 47:152–161
Zhang RL, Zhang B (2014) Prospects of using DNA barcoding for species identification and evaluation of the accuracy of sequence databases for ticks (Acari: Ixodida). Ticks Tick Borne Dis 3:352–358
Acknowledgements
This study was financially supported by the National Key Research and Development Program of China (2016YFC1202000, 2016YFC1202002); NSFC (31572511, 31471967, 31402189); 973 Program (2015CB150300); ASTIP, FRIP (2014ZL010), CAAS; NBCIS CARS-38; Jiangsu Co-innovation Center program for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, State Key Laboratory of Veterinary Etiological Biology Project.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Liu, X., Chen, Z., Ren, Q. et al. Genetic diversity of Haemaphysalis qinghaiensis (Acari: Ixodidae) in western China. Exp Appl Acarol 74, 427–441 (2018). https://doi.org/10.1007/s10493-018-0242-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-018-0242-2