
Abstract
An orange pigmented, Gram-staining negative, aerobic, motile, rod-shaped bacterium isolated from a soil from the Tanggula Mountain, China was studied using a polyphasic approach. Based on 16S rRNA gene sequence similarity, strain W16RDT was found to be closely related to Sphingomonas prati DSM 103336T (99%), Sphingomonas fennica DSM 13665T (97.21%), followed by Sphingomonas laterariae DSM 25432T (96.44%), Sphingomonas haloaromaticamans CGMCC 1.10206 T (96.36%) and Sphingomonas formosensis DSM 24164T (96.06%). The strain was found to be catalase and oxidase positive and was found to grow optimally at temperatures of 20–25 °C, pH 8 and tolerated NaCl concentration up to 1% (w/v). The major fatty acids identified were summed feature eight comprising C18:1 ω 7c and/or C18:1 ω 6c (39.2%), summed feature three comprising of C16:1 ω7c and/or C16:1 ω6c (36.7%) and C16:0 (7.0%). The polar lipids detected were phosphatidylcholine, sphingoglycolipid, phosphatidylglycerol, phosphatidylethanolamine, diphosphatidylglycerol, phosphatidyldimethylethanolamine, phosphatidylmonomethylethanolamine, and three unidentified lipids. The strain possessed ubiquinone-10 (Q-10) as the predominant respiratory quinone. Along with other distinguishing characteristics, we also describe the draft genome of strain W16RDT. The final assembled draft genome sequence is 3,722,743 bp with 3390 coding and 48 RNA (45 tRNA and 3 rRNA) genes. The DNA G+C content of the genomic DNA was determined to be 67%. The DNA–DNA relatedness value between the strain W16RDT and its closest phylogenetic relatives S. prati DSM 103336T, S. fennica DSM 13665T, S. laterariae DSM 25432T, and S. haloaromaticamans CGMCC 1.10206T were 52.17, 47.60, 20.93 and 17.09% respectively. The strain W16RDT could be distinguished genotypically and phenotypically from the recognized species belonging to the genus Sphingomonas and thus represents a novel species, for which the name Sphingomonas montana sp. nov. is proposed. The type strain is W16RDT (=CGMCC 1.15646T = DSM 103337T).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Sphingomonas, which belongs to Alphaproteobacteria (Anzai et al. 2000), was originally described by Yabuuchi et al. (1990) taking Sphingomonas paucimobilis as the type species. Later Takeuchi et al. (2001) reclassified the genus Sphingomonas into four separate genera, as Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis on the basis of phylogenetic, chemotaxonomic and phenotypic characteristics. The genus has been subsequently emended by others (Busse et al. 2003; Chen et al. 2012). Members of the genus Sphingomonas are Gram-negative, strictly aerobic, chemoheterotrophic, non-spore forming, and either motile or non-motile rods which can develop into off white, yellow or orange colored colonies. Chemotaxonomically they are distinguished by the presence of ubiquinone 10 (Q-10) and 2-hydroxy fatty acids, as well as the absence of 3-hydroxy fatty acids (Zhang et al. 2005), and they contain sphingoglycolipids as major components of their outer membrane. Species of the genus Sphingomonas share a wide range of environmental habitats such as soils, air (Kim et al. 2014; Lee et al. 2015; Margesin et al. 2012), ice (Liu et al. 2015), plants (Huang et al. 2012), the phyllosphere (Talà et al. 2013), marine bivalves (Romanenko et al. 2007) and ground water (Wittich et al. 2007). The genus Sphingomonas has been exploited for many biotechnological applications, such as the bioremediation of environmental contaminants (Coppotelli et al. 2008), or the production of extracellular polymers (Pollock 1993). At the time of writing, the genus Sphingomonas comprises of 95 recognized species (http://www.bacterio.net/sphingomonas.html). Here, we report on the taxonomic characterization of a bacterial strain designated as W16RDT which was isolated from a soil sample collected from the Tanggula Mountain in the Qinghai- Tibetan Plateau. Based on the phenotypic, phylogenetic, chemotaxonomic and genotypic results, strain W16RDT represents a novel species of the genus Sphingomonas.
Materials and methods
Isolation of bacterium and culture conditions
In a study carried out to explore the diversity of culturable soil bacteria from different regions of the Tibetan Plateau, strain W16RDT was isolated from a soil from the Tanggula Mountain, China. The soil sample was diluted with R2A broth and spread on R2A agar (Difco, USA) plates that were incubated at 10 °C for 4–7 days. The colonies developed were restreaked several times on R2A agar to obtain a pure culture. A single colony of the isolate W16RDT was transferred to a fresh R2A agar plate and incubated at 25 °C, followed by routine subculturing. Purity was confirmed by microscopic examination. The strain was then stored at −80 °C in glycerol (15%, v/v). The strain has been deposited to the China General Microbiological Culture Collection Center (CGMCC 1.15646T) and German Collection of Microorganisms and Cell cultures (DSM 103337T). Reference strains, Sphingomonas prati DSM 103336T, Sphingomonas fennica DSM 13665T, Sphingomonas laterariae DSM 25432T and Sphingomonas formosensis DSM 24164T, were obtained from DSMZ (German Collection of Microorganisms and Cell cultures), and Sphingomonas haloaromaticamans CGMCC 1.10206T was obtained from CGMCC (China General Microbiological Culture Collection Center). All reference strains were cultured in parallel with the strain W16RDT under the same conditions throughout all experiments.
Phenotypic and biochemical characteristics
Cell morphology and the presence of flagella were determined by transmission electron microscopy (JEM-1400; JEOL) with fresh cells at their exponential phase of growth on R2A agar. The motility test was carried out by stab inoculation of the bacterium on R2A medium with 0.5% agar (Gu et al. 2015). Gram staining was performed following the method of Smibert and Krieg (1994). Oxidase activity was examined by using an oxidase reagent (bioMerieux) and catalase activity was interpreted by the production of oxygen bubbles in 3% (v/v) aqueous hydrogen peroxide solution. Optimum conditions for the growth including temperature (0, 4, 10, 15, 20, 25, 30 and 35 °C), pH (4.0–11.0 at interval of 1.0 pH unit) and NaCl concentration [(0, 1, 2, 3, 4, 5 and 6% (w/v)] were determined on R2A broth over an incubation period of 7 days at 25 °C. The pH values were adjusted using the buffers 0.1 M citric acid/0.1 M sodium citrate (for the pH 4.0–5.0), 0.1 M KH2PO4/0.1 M NaOH (for the pH 6.0-8.0), 0.1 M NaHCO3/0.1 M Na2CO3 (for the pH 9.0–10.0) and 0.05 M Na2HPO4/0.1 M NaOH (for the pH 11.0) (Zhu et al. 2015) and different concentrations of NaCl were obtained by supplementing the growth medium with 1–6% (w/v at interval of 1% unit) NaCl. Other physiological and biochemical properties were determined with API 20NE and API ZYM strips following the instructions provided by the manufacturer (bioMerieux,).
DNA extraction, PCR amplification, and whole genome sequencing
The genomic DNA was extracted according to the method described by Marmur (1961) and its purity was assessed using a Nano-Drop spectrophotometer (2000c; Thermo). The universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-CGGTTACCTTGTTACGACTT-3′) were used to amplify the 16S rRNA gene from strain W16RDT (Embley 1991) and the purified PCR products were sequenced by Sangon Biotech (Beijing). The 16S rRNA gene sequence of the strain was then compared with the available sequences in the EzTaxon-e-server (http://eztaxon-e.ezbiocloud.net) (Kim et al. 2012) and GenBank using BLAST program (NCBI) in order to determine its phylogenetic affiliation.
The strain was further subjected to whole genome sequencing, for which genomic DNA of the bacterium was extracted using a TIANamp bacterial DNA kit (Tiangen Biotech (Beijing) Co. Ltd) according to the manufacturer’s instructions. In brief, a single colony from the pure culture of the strain W16RDT was cultured in R2A broth tubes at the temperature of 25 °C until abundant growth was observed and then subjected to DNA extraction. The extracted DNA was prepared for whole-genome sequencing, which was performed at the Microbial Genome Research Center, Institute of Microbiology, Chinese Academy of Sciences, Beijing. In brief, the genomic DNA was fragmented by ultrasonication, and the DNA fragments were subjected to the whole-genome sequencing workflow of the Illumina HiSeq 2000 system.
Genome assembly and annotation
Genome assembly was performed by SOAPdenovo (http://soap.genomics.org.cn) and the gaps were closed by using SOAP GapCloser (http://soap.genomics.org.cn). Glimer 3.02 (Delcher et al. 2007) served for the purpose of the prediction of open reading frame and tRNAscan-SE and RNAmmer (Lagesen et al. 2007) were used for the identification of tRNA and rRNA respectively. The annotation of the genome sequences was done with the help of the RAST program (Rapid Annotation using Subsystem Technology) (Aziz et al. 2008). The annotation results were then checked and compared with the database of NCBI-NR (http://www.ncbi.nlm.nih.gov/), COG (Tatusov et al. 2003).
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MOLY00000000. The version described in this paper is version MOLY01000000.
Comparative genomics
For the comparative analysis, genome sequences of 47 previously reported isolates (GenBank accession number NZ_MIPT01000001, NZ_FUYM01000001, NZ_LDTD01000060, NZ_CAVK010000039, NC_014006, NZ_LDTB01000010, NZ_LQQO01000001, NZ_CP010836, NZ_LT840185, NZ_CP018820, NZ_BBJS01000014, NZ_LDUA01000001, NZ_JFYY01000001, NZ_LYMJ01000001, FOXP01000001, NZ_CP009571, NZ_LDTF01000007, NC_008048, NZ_BCTR01000001, NZ_CP014168, NZ_JH584235, NZ_AGFU01000002, NZ_KK073876, NZ_AQUJ01000001, NZ_KB900605, NZ_ATTG01000001, NZ_ATTI01000001, NZ_KE386571, NZ_CP006644, NZ_AHKO01000024, NC_009511, NZ_BCWT01000001, NZ_BCYU01000001, NZ_BBWU01000048, BCYW01000001, NZ_BCYX01000001, NZ_LQCK02000001, NZ_BBPI01000034, NZ_BCYY01000001, NZ_BCYZ01000002, NZ_BCTY01000001, NZ_LDTC01000066, NZ_LDTE01000010, BCTZ01000004, NZ_JXTP01000018, NZ_JQMC01000001, NZ_JONN01000001) were downloaded from the NCBI website and the average nucleotide identity between these genomes and the genome of the type strain W16RDT was calculated based on fragmented alignments by using a Perl script.
Phylogenetic analysis
The 16S rRNA gene sequence (1483 bp) was extracted from the draft genome sequence of strain W16RDT, and it was compared with the available sequences in EzTaxon-e-server (http://eztaxon-e.ezbiocloud.net) and GenBank using BLAST program (NCBI) to determine its phylogenetic affiliation. The 16S rRNA gene sequences were then analyzed with the software package MEGA 5.05 (Tamura et al. 2011). The phylogenetic tree was constructed using the neighbor-joining method with bootstrap values based on 1000 replications.
Cellular fatty acids, polyamines, isoprenoid quinones and polar lipid analysis
For the analysis of cellular fatty acids, strain W16RDT and the five reference strains were grown on R2A agar at 25 °C for 2–4 days, and cells were collected at their exponential phase of growth. The extraction and analysis of fatty acid methyl esters were performed according to the standard protocol of the Microbial Identification System (MIDI, version 6.0). The standardization of the physiological age of strain W16RDT and its reference strains was done following the protocol by MIDI (http://www.microbialid.com/PDF/TechNote_101.pdf). For the determination of isoprenoid quinone and polar lipids, cells were harvested after 72 h of growth at 25 °C. Isoprenoid quinones were analyzed as per the method described by Hiraishi et al. (1998) by using a Waters Acquity Ultra Performance LC (UPLC)-Q-TOF–MS spectrometer in electrospraying ionization mode (Romano et al. 2006). The extraction and analysis of polar lipids were performed by two-dimensional TLC (Altenburgera et al. 1996; Tindall 1990). Total lipid material and specific functional groups were detected by using molybdatophosphoric acid (total lipids), molybdenum blue (phosphor group), ninhydrin (amino group), and α-naphthol (glycolipid). To determine the polyamine content of the strain W16RDT, the draft genome of the strain was compared with the spermidine synthase gene [key enzyme responsible for spermidine biosynthesis (Lee et al. 2009)] of Sphingomonas wittichii RW1 using BLAST (Altschul et al. 1990). All the proteins of the strain W16RDT and homospermidine synthase proteins of S. wittichii RW1 were compared against the Pfam-A database version 27.0 (Finn et al. 2014) using the HMMER 3.0 software (Eddy 2008).
DNA–DNA hybridisation and determination of DNA G+C content
DNA–DNA hybridisation experiments were carried out between strain W16RDT and four reference strains, S. prati DSM 103336T, S. fennica DSM 13665T, S. laterariae DSM 25432T and S. haloaromaticamans CGMCC 1.10206 T. The hybridisation experiment was performed as described by Ley et al. (1970) and Huss et al. (1983). The mean (±SD) DNA reassociation rate between the strains was calculated taking data from three independent experiments.
The DNA G+C content percentage of the strain W16RDT was calculated from the draft genome sequences as (G+C)/(A+T+C+G) by using a perl script.
Results and discussion
Morphological and phenotypic characteristics
Colonies of strain W16RDT were seen to be orange, circular, smooth, flat and transparent on R2A agar plates after incubation for 3–4 days at 25 °C. The cells were found to be rod-shaped, motile by single polar flagellum and measured 1.2 µm in length and 0.91 µm in diameter (Figure S1). The strain was found to grow in the temperature range 4–30 °C (optimum, 20–25 °C) and pH range 5–9 (optimum, 8) and to be tolerant of NaCl concentration up to 1% (w/v) and catalase and oxidase positive. In the APIZYM system, the strain was positive for esterase lipase (C8), ß-galactosidase, ß-glucosidase, alkaline phosphatase, esterase (C4), leucine arylamidase, acid phosphatase and weakly positive for valine arylamidase, naphthol-AS-BI-phosphohydrolase, α–galactosidase and in the API 20NE system it was positive for para-nitrophenyl- βD-galactopyranosidase, esculin hydrolysis and assimilation of L-arabinose. A number of differentiating characteristics that can be used to distinguish the strain W16RDT from other recognized species of the genus Sphingomonas are displayed in Table 1.
Genome properties and annotation
The genome of strain W16RDT produced a total of 6,617,100 raw reads with a mean read length of 125 bp. After assembling, a total of 35 scaffolds with N50 length of 20,22,37 bp and maximum length of 425,724 bp were obtained. The completed assembled draft genome is 37,22,743 bp. Of the 3438 predicted genes, 3390 were protein coding genes and 48 RNA (45 tRNA genes and 3 rRNA) genes. Rast annotation of the whole genome showed the presence of 2654 COG categories. Basic sequence statistics of the draft genome of the strain W16RDT are shown in Table 3.
Comparative genomics
The ANI comparison between the genome of the strain W16RDT with that of the published genomes of related species (Figure S3) showed results less than 95% (the species ANI cut off value), clearly indicating that the strain W61RDT belongs to a new species.
Phylogenetic analysis
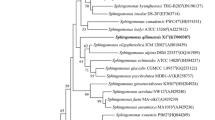
The 16S rRNA gene sequence of the strain W16RDT obtained in this study was a continuous stretch of 1483 bp and has been deposited in the GenBank database (accession number KU535674). Comparison of the 16S rRNA gene sequence of strain W16RDT with the available sequences in EzTaxon-e-server (http://eztaxon-e.ezbiocloud.net) and GenBank (http://www.ncbi.nlm.nih.gov) revealed the strain to be closely related to S. prati DSM 103336T (99%) followed by S. fennica DSM 13665T (97.21%), S. laterariae DSM 25432T (96.44%), S. haloaromaticamans CGMCC 1.10206 T (96.36%) and Sphingomonas formosensis DSM 24164T (96.06%). The relationship between the stain W16RDT and members of the genus Sphingomonas was further evident in a neighbor joining phylogenetic tree based on 16S rRNA gene sequences, which positioned the strain W16RDT within a clade comprising species of the genus Sphingomonas (Fig. 1). Similar topologies were recovered using the maximum-parsimony and maximum- likelihood algorithms (data not shown) supporting the phylogenetic position of strain W16RDT within the genus Sphingomonas.

Neighbour-joining phylogenetic tree based on 16S rRNA gene sequences showing the position of strains W16RDT in relation to the type strains of Sphingomonas species and Novosphingobium stygium strain IFO 16085T and Polymorphobacter multimanifer 262-7T was used as an outgroup. Filled circles and filled triangles indicate that the corresponding nodes were also recovered in the trees generated with the maximum- likelihood and maximum-parsimony algorithms respectively. Bootstap percentages were based on 1000 replications. Bar 0.01 substitutions per nucleotide position
Cellular fatty acids, isoprenoid quinones, polar lipid and polyamines analysis
The predominant fatty acids in the strain W16RDT were found to be summed feature eight comprising C18:1 ω 7c and/or C18:1 ω 6c (39.2%), summed feature three comprising of C16:1 ω7c and/or C16:1 ω 6c (36.7%) and C16:0 (7.0%).The relative differences in the respective proportions of C16:1 2-OH, C18:1 2-OH, C16:1 ω5C, C17:1 ω6c and a high percentage of C16:1 ω 7c and/or C16:1 ω 6c distinguish the strain W16RDT from its close relatives considered in this study (Table 2). Ubiquinone 10 (Q-10) was determined to be the major respiratory quinone and the polar lipids detected were phosphatidylcholine (PC), sphingoglycolipid (SGL), phosphatidylglycerol (PG), phosphatidylethanolamine (PE), diphosphatidylglycerol (DPG), phosphatidyldimethylethanolamine (PDE), phosphatidylmonomethylethanolamine (PME) and three unidentified lipids, L1-L3 (Figure S2.a, A1). The presence of phosphatidyldimethylethanolamine (PDE) in strain W16RDT made it distinct from its close relative S. prati. Also, the strain W16RDT showed the presence of three unidentified lipids (L1-L3) in contrast to the S. prati type strain which showed the presence of only two unidentified lipids, L1-L2 (Fig S2.a, A1/B1). However, the presence of PDE in the strain W16RDT was unique amongst its closely related reference strains considered in this study (Figure S2.a, S2.d). The polar lipid profile of strain W16RDT and the reference strains is presented in Table S1. One protein of strain W16RDT showed 56% amino acid identity to spermidine synthase protein in S. wittichii RW1 but no protein showed any match to homospermidine synthase. We also compared all proteins of the strain to the Pfam database, but did not find any protein containing the domain of homospemidine synthase protein. Thus strain W16RDT is predicted to possess spermidine as its polyamine, as has been reported in many species of the genus Sphingomonas, such as S. lacus, S. aquatilis, S. koreensis, S. cloacae, S. wittichii and S. abikonensis (Hamana et al. 2003; Kim et al. 2015; Yabuuchi and Kosako 2005).
DNA–DNA hybridisation and DNA G+C content
The mean (± SD) DNA reassociation rate between the strain W16RDT and S. prati DSM 103336T, S. fennica DSM 13665T, S. laterariae DSM 25432T, and S. haloaromaticamans CGMCCT were 52.17, 47.60, 20.93 and 17.09% respectively (Table 1) and the DNA G + C content of the genomic DNA was determined to be 67%.
Taxonomic conclusion
On the basis of 16S rRNA gene sequence analysis, strain W16RDT showed high sequence similarity with S. prati DSM 103336T (99%) followed by S. fennica DSM 13665T (97.21%), S. laterariae DSM 25432T (96.44%), S. haloaromaticamans CGMCC 1.10206T (96.36%) and S. formosensis DSM 24164T (96.06%). The phylogenetic affiliation of the strain with the genus Sphingomonas is supported by its position in the neighbor-joining tree where it clustered with the species of the genus Sphingomonas (Fig. 1). Distinct phenotypic characteristics, a low level of DNA–DNA hybridisation values (Table 1) and a low ANI value of <95%, (Figure S3) justify assignment of strain W16RDT to the genus Sphingomonas as the type strain of a novel species, for which the name Sphingomonas montana sp. nov. is proposed.
Description of Sphingomonas montana sp. nov.
Sphingomonas montana (mon.ta’na. L. fem. adj. montana belonging to a mountain).
Colonies on R2A agar are round, smooth, opaque, flat and orange colored. Cells are Gram-staining negative, rod-shaped, motile by single polar flagellum and 1.2 µm in length and 0.9 µm in diameter. Growth in R2A broth occurs at the temperature range 4–30 °C (optimum, 20–25 °C), pH 5–9 (optimum, 8) and with 0–1% (w/v) NaCl (optimum 0%). The major fatty acids are summed feature eight comprising C18:1 ω 7c and/or C18:1 ω 6c, summed feature three comprising of C16:1 ω 7c and/or C16:1 ω 6c and C16:0 and Q-10 is the major respiratory quinone. The G + C content of the genomic DNA of the type strain is 67% and the final assembled draft genome sequence of the type strain is 3,722,743 bp. The polar lipids are phosphatidylcholine, sphingoglycolipid, phosphatidylglycerol, phosphatidylethanolamine, diphosphatidylglycerol, phosphatidyldimethylethanolamine, phosphatidylmonomethylethanolamine and three unidentified lipids. The type strain is W16RDT (=CGMCC 1.15646T=DSM 103337T) isolated from soil from the Tangulla Mountain, China. The Genbank accession number for the 16S rRNA gene sequence and genome of the strain is KU535674 and MOLY00000000 respectively. The DPD TaxonNumber is TA00189.
References
Altenburgera P, Kämpferb P, Makristathisc A, Lubitza W, Bussea H-J (1996) Classification of bacteria isolated from a medieval wall painting. J Biotechnol 47:39–52
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. doi:10.1016/S0022-2836(05)80360-2
Anzai Y, Kim H, Park J-Y, Wakabayashi H, Oyaizu H (2000) Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int J Syst Evol Microbiol 50:1563–1589
Aziz RK et al (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genom 9:75
Busse HJ, Denner EB, Buczolits S, Salkinoja-Salonen M, Bennasar A, Kampfer P (2003) Sphingomonas aurantiaca sp. nov., Sphingomonas aerolata sp. nov. and Sphingomonas faeni sp. nov., air- and dustborne and Antarctic, orange-pigmented, psychrotolerant bacteria, and emended description of the genus Sphingomonas. Int J Syst Evol Microbiol 53:1253–1260. doi:10.1099/ijs.0.02461-0
Chen H et al (2012) Reclassification and emended description of Caulobacter leidyi as Sphingomonas leidyi comb. nov., and emendation of the genus Sphingomonas. Int J Syst Evol Microbiol 62:2835–2843
Coppotelli B, Ibarrolaza A, Del Panno M, Morelli I (2008) Effects of the inoculant strain Sphingomonas paucimobilis 20006FA on soil bacterial community and biodegradation in phenanthrene-contaminated soil. Microb Ecol 55:173–183
Delcher AL, Bratke KA, Powers EC, Salzberg SL (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679
Eddy SR (2008) A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput Biol 4:e1000069. doi:10.1371/journal.pcbi.1000069
Embley TM (1991) The linear PCR reaction: a simple and robust method for sequencing amplified rRNA genes. Lett Appl Microbiol 13:171–174
Finn RD et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–230. doi:10.1093/nar/gkt1223
Gu Z et al (2015) Hafnia psychrotolerans sp. nov., isolated from lake water. Int J Syst Evol Microbiol 65:971–974. doi:10.1099/ijs.0.000049
Hamana K, Sakamoto A, Tachiyanagi S, Terauchi E (2003) Polyamine profiles of some members of the gamma subclass of the class Proteobacteria: polyamine analysis of twelve recently described genera. Microbiol Cult Collect 19:3–11
Hiraishi A, Ueda Y, Ishihara J (1998) Quinone profiling of bacterial communities in natural and synthetic sewage activated sludge for enhanced phosphate removal. Appl Environ Microb 64:992–998
Huang H-Y et al (2012) Sphingomonas endophytica sp. nov., isolated from Artemisia annua L. Int J Syst Evol Microbiol 62:1576–1580
Huss VA, Festl H, Schleifer KH (1983) Studies on the spectrophotometric determination of DNA hybridization from renaturation rates. Syst Appl Microbio 4:184–192
Kaur J, Kaur J, Niharika N, Lal R (2012) Sphingomonas laterariae sp. nov., isolated from a hexachlorocyclohexane-contaminated dump site. Int J Syst Evol Microbiol 62:2891–2896
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S, Chun J (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721
Kim SJ, Moon JY, Lim JM, Ahn JH, Weon HY, Ahn TY, Kwon SW (2014) Sphingomonas aerophila sp. nov. and Sphingomonas naasensis sp. nov., isolated from air and soil, respectively. Int J Syst Evol Microbiol 64:926–932. doi:10.1099/ijs.0.055269-0
Kim JH, Kim SH, Kim KH, Lee PC (2015) Sphingomonas lacus sp. nov., an astaxanthin-dideoxyglycoside-producing species isolated from soil near a pond in South Korea. Int J Syst Evol Microbiol 65(9):2824–2830
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108
Lee J, Sperandio V, Frantz DE, Longgood J, Camilli A, Phillips MA, Michael AJ (2009) An alternative polyamine biosynthetic pathway is widespread in bacteria and essential for biofilm formation in Vibrio cholerae. J Biol Chem 284:9899–9907
Lee KC, Kim KK, Kim J-S, Kim D-S, Ko S-H, Yang S-H, Lee J-S (2015) Sphingomonas vulcanisoli sp. nov., isolated from soil of a lava forest. Int J Syst Evol Microbiol 65:3320–3325
Ley Jd, Cattoir H, Reynaerts A (1970) The quantitative measurement of DNA hybridization from renaturation rates. Eur J Bioche 12:133–142
Lin S-Y et al (2012) Sphingomonas formosensis sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from agricultural soil. Int J Syst Evol Microbiol 62:1581–1586
Liu Q, Liu H-C, Zhang J-L, Zhou Y-G, Xin Y-H (2015) Sphingomonas psychrolutea sp. nov., a psychrotolerant bacterium isolated from glacier ice. Int J Syst Evol Microbiol 65:2955–2959
Manandhar P, Zhang G, Hu Y, Lama A, Gao F, Gu Z (2016) Sphingomonas prati sp. nov., isolated from alpine meadow soil. Int J Syst Evol Microbiol 66:4269–4275
Margesin R, Zhang D-C, Busse H-J (2012) Sphingomonas alpina sp. nov., a psychrophilic bacterium isolated from alpine soil. Int J Syst Evol Microbiol 62:1558–1563
Marmur J (1961) A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol 3:208
Pollock TJ (1993) Gellan-related polysaccharides and the genus Sphingomonas. Microbiology 139:1939–1945
Romanenko LA, Uchino M, Frolova GM, Tanaka N, Kalinovskaya NI, Latyshev N, Mikhailov VV (2007) Sphingomonas molluscorum sp. nov., a novel marine isolate with antimicrobial activity. Int J Syst Evol Microbiol 57:358–363
Romano I, Lama L, Nicolaus B, Poli A, Gambacorta A, Giordano A (2006) Halomonas alkaliphila sp. nov., a novel halotolerant alkaliphilic bacterium isolated from a salt pool in Campania (Italy). J Gen Appl Microbiol 52:339–348
Smitbert RM, Krieg NR (1994) Phenotypic characterization. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR (eds) Methods for general and molecular bacteriology. American Society for Microbiology, Washington, DC, pp 607–654
Takeuchi M, Hamana K, Hiraishi A (2001) Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int J Syst Evol Microbiol 51:1405–1417
Talà A et al (2013) Sphingomonas cynarae sp. nov., a proteobacterium that produces an unusual type of sphingan. Int J Syst Evol Microbiol 63:72–79
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tindall BJ (1990) Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol Lett 66:199–202
Wittich RM, Busse HJ, Kampfer P, Macedo AJ, Tiirola M, Wieser M, Abraham WR (2007) Sphingomonas fennica sp. nov. and Sphingomonas haloaromaticamans sp. nov., outliers of the genus Sphingomonas. Int J Syst Evol Microbiol 57:1740–1746
Yabuuchi E, Kosako Y (2005) Order IV. Sphingomonadales ord. nov. In: Brenner DJ, Kreig NR, Staley JT, Garrity GM (eds) Bergey’s manual of systematic bacteriology, vol 2, 2nd edn. Springer, New York, pp 230–233
Yabuuchi E, Yano I, Oyaizu H, Hashimoto Y, Ezaki T, Yamamoto H (1990) Proposals of Sphingomonas paucimobilis gen. nov. and comb. nov., Sphingomonas parapaucimobilis sp. nov., Sphingomonas yanoikuyae sp. nov., Sphingomonas adhaesiva sp. nov., Sphingomonas capsulata comb. nov., and two genospecies of the genus Sphingomonas. Microbiol Immunol 34:99–119
Zhang Y-Q, Chen Y-G, Li W-J, Tian X-P, Xu L-H, Jiang C-L (2005) Sphingomonas yunnanensis sp. nov., a novel Gram-negative bacterium from a contaminated plate. Int J Syst Evol Microbiol 55:2361–2364
Zhu L et al (2015) Sphingomonas gei sp. nov., isolated from roots of Geum aleppicum. Int J Syst Evol Microbiol 65:1160–1166
Acknowledgements
This work was supported by the key project from National Science and Foundation of China (31290222), project from National Science Foundation of China (41172307) and the 973 Project from the Science & Technology Department of China (2013CB956002). We would like to express our gratitude to Dr. Manik Ghosh and Dr. Amy for their technical help and Mrs. Yanhua Sun, Dr. Liang Shen, and Zhengquan Gu for their suggestions during the experimental work. We thank Prof. Aharon Oren for assistance with nomenclature.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
The Genbank accession number for the 16S rRNA gene sequence of strain W16RDT is KU535674.
The Genbank accession number for the whole genome sequence of the strain W16RDT is MOLY00000000.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Manandhar, P., Zhang, G., Lama, A. et al. Sphingomonas montana sp. nov., isolated from a soil sample from the Tanggula Mountain in the Qinghai Tibetan Plateau. Antonie van Leeuwenhoek 110, 1659–1668 (2017). https://doi.org/10.1007/s10482-017-0915-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-017-0915-6