
Abstract
The interaction of keto-acids with reactive oxygen species (ROS) is known to produce the corresponding carboxylic acid with the concomitant formation of CO2. Formate is liberated when the keto-acid glyoxylate neutralizes ROS. Here we report on how formate is involved in combating oxidative stress in the nutritionally-versatile Pseudomonas fluorescens. When the microbe was subjected to hydrogen peroxide (H2O2), the levels of formate were 8 and two-fold higher in the spent fluid and the soluble cell-free extracts obtained in the stressed cultures compared to the controls respectively. Formate was subsequently utilized as a reducing force to generate NADPH and succinate. The former is mediated by formate dehydrogenase (FDH-NADP), whose activity was enhanced in the stressed cells. Fumarate reductase that catalyzes the conversion of fumarate into succinate was also markedly increased in the stressed cells. These enzymes were modulated by H2O2. While the stressed whole cells produced copious amounts of formate in the presence of glycine, the cell-free extracts synthesized ATP and succinate from formate. Although the exact role of formate in anti-oxidative defence has to await further investigation, the data in this report suggest that this carboxylic acid may be a potent reductive force against oxidative stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As oxidative stress is part of aerobic life, most organisms have evolved intricate mechanisms to circumvent this challenge. These strategies include the utilization of enzymes such as catalase, superoxide dismutase (SOD) and glutathione peroxidase which aid in the lowering of oxidative tension during aerobic respiration (Brioukhanov et al. 2006). Recently the significance of ketoacids in combatting reactive oxygen species (ROS) has emerged. The moieties such as alpha ketoglutarate (AKG), oxaloacetate (OAA), pyruvate and glyoxylate scavenge ROS with the concomitant formation of their respective carboxylic acid (Alhasawi et al. 2015a; Lemire and Appanna 2011; Mailloux et al. 2008; Singh et al. 2008; Li et al. 2009; Thomas et al. 2015). When glyoxylate is utilized as an ROS scavenger, formate is one of the critical by-products formed (Alhasawi et al. 2015b; Yokota et al. 1983; Yokota et al. 1985). In numerous bacteria, plants and animals formate is an important metabolite involved in energy metabolism (Hourton-Cabassa et al. 1998; Leonharsberger et al. 2002). Its favourable redox potential enables this monocarboxylic acid to be oxidised not only through the aerobic respiratory pathways but to serve as an electron donor for the reduction of key metabolites such as fumarate, nitrate and nitrite (Bagramyan et al. 2000; Jormakka et al. 2002; Shinagawa et al. 2008; Su and Puls 2004).
Formate may undergo different fates in these bacteria depending on the physiological conditions (Leonharsberger et al. 2002). It may be secreted, oxidised aerobically, used as a reductant or converted into CO2 and H2 via the enzyme formate hydrogen lyase (Bagramyan and Trchounian 2003; Yoshida et al. 2005). A key enzyme in the metabolism of formate is formate dehydrogenase (FDH). In some bacteria this enzyme predominantly utilizes nitrate as an electron acceptor (Uchimura et al. 2002). FDH plays a pivotal role in respiration as well as in the maintenance of a reducing environment (Jormakka et al. 2002). These enzymes have been reported with differing cofactor requirements, electron acceptors, substrates and cellular locations (Hourton-Cabassa et al. 1998). The FDHs NAD-dependent which have been extensively studied in both bacteria, yeast and plants, have been widely utilized in industry for NADH regeneration (Alekseeva et al. 2011; Hoelsch et al. 2013; Suzuki et al. 1998).The occurrence of FDH NADP-dependent has also been reported (Yammamoto et al. 1983; Gul-Karaguler et al. 2001). FDH synthesis has been shown to increase strongly under conditions of stress including abrupt changes in temperature, irradiation with UV light, hypoxia and chemical agents and may contribute in maintaining NADPH homeostasis (Andreadeli et al. 2009; Andreadeli et al. 2009; Hoelsch et al. 2013; Hourton-Cabassa et al. 1998; Jormakka et al. 2003).
The role of this monocarboxylic acid in providing the reducing power to the pivotal ribonucleotide reductase has been shown (Stubbe et al. 2003). This enzyme participates in the synthesis of deoxyribonucleotides with the concomitant formation of CO2. Deoxyribonucleotides are critical in the cellular replication and their formation usually necessitates the utilization of NADPH. As part of our study to unravel the significance of metabolic pathways in anti-oxidative defence, we have evaluated the role of formate in the adaptation of the nutritionally-versatile microbe Pseudomonas fluorescens to oxidative stress. Here we report that the presence of the enhanced level of this mono- carbon carboxylic acid is an important source of reducing power in the H2O2-challenged cells. It participates in the synthesis of NADPH and contributes to the reduction of fumarate to succinate, biological reactions that ensure the survival of the microbe in an oxidative milieu. The importance of formate as a substitute for NADH in environments where the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OP) are ineffective is also discussed.
Methods
Growth
P. fluorescens from American type culture collection (ATCC13525) was grown in defined citrate/glycine media (control), consisting of Na2HPO4 (6 g), KH2PO4 (3 g), 15 mM glycine (1.2 g), MgSO4⋅7H2O (0.2 g), and 19 mM citrate (4 g) with a pH of 6.8. Additionally, trace elements were added as descri in (Mailloux et al. 2008). Media were dispensed into 200 mL aliquots in two 500 mL Erlenmeyer flasks (control and stress conditions) and autoclaved for 20 min at 121 °C prior to the inoculation with 1 ml of bacteria grown to stationary phase in a control medium (same conditions as control culture from the experiment). Oxidative stress in the stressed culture was introduced by the addition of 500 µM H2O2, an amount known to elicit maximal antioxidative response (Alhasawi et al. 2015a, b). All cultures were aerated in a gyratory water bath shaker, model 76 (New Brunswick Scientific) at 26 °C at 140 rpm. The cells and spent fluid were isolated at the stationary phase of growth for metabolomic and enzymatic analyses (28 h for control and 50 h growth for the H2O2 stressed cultures). Following the harvesting of cells at various growth intervals, the bacterial pellets were treated with 0.5 N NaOH and cell growth was monitored by measuring the solubilized protein using the Bradford assay (Bradford 1976).
Regulation and whole cell experiments
In the regulation experiments, to assess the adaptive and reversible nature of the shifts in metabolism, cells were harvested at stationary phase of growth. Control cells (10 mg protein equivalent) were incubated for 8 h in 50 mL media containing 500 µM H2O2 whereas the H2O2-stressed cells were incubated for 8 h in 50 mL control media as described in (Alhasawi et al. 2014). The cell-free extracts and the spent fluid were subsequently analyzed for metabolites and enzymatic activities.
To evaluate the source of formate production, whole cells (10 mg protein equivalent) from the control and stress cultures were incubated for 8 h in separate media containing the same growth nutrients as the control culture but with only citrate or glycine in the presence and in the absence of H2O2. To monitor the rate of formate utilization, 10 mg protein equivalent of control and stressed whole cells were incubated in reaction mixture containing 5 mM formate and the consumption of the monocarboxylic acid was recorded by HPLC after 8 h.
Cell fractionation
Following the isolation of the bacteria at 4 °C for 10 min at 10,000 g with the aid of a Sorvall Legend RT Centrifuge, cells were washed with 0.85 % NaCl and respun before being resuspended in 500 µL cell storage buffer (CSB) consisting of 50 mM Tris–HCl, 5 mM MgCl2 and 1 mM fluoride (PMSF). Sonication was utilized to lyse the cells (unbroken cells were removed by centrifugation at 10,000 g). These were centrifuged at 180,000 g for 3 h at 4 °C yielding a soluble cell-free (CFE) and a membrane fraction. The membrane fraction was suspended in 500 µL of CSB. The Bradford assay was utilized to determine protein content with serum bovine albumin as the standard. Equal protein concentrations were utilized in all experiments.
Enzymatic studies
BN-PAGE was executed as per the protocol described in, Auger et al. (2015); Mailloux et al. (2008) and Schagger and von Jagow (1991). For these assays, a 4–16 % gradient gel was prepared and the protein (4 μg/μL) was prepared in blue native buffer (400 mM 6-amino hexanoic acid, 50 mM Bis–Tris [pH 7.0]). To solubilize membrane bound proteins in order to ensure optimal protein separation, a final concentration of 1 % dodecyl-β-maltoside was added to the membrane fractions. Protein samples were loaded into each well of the native gel (10–60 µg) and electrophoresed at 4 °C under native conditions at 80 V and 15 mA for proper stacking followed by 150 V and 25 mA in the resolving gel for the migration of the protein until it travelled half-way through the gel. At the halfway point, blue cathode buffer (50 mM Tricine, 15 mM Bis–Tris, 0.02 % w/v Coomassie G-250, pH 7 at 4 °C) was changed to a colorless cathode buffer (50 mM Tricine, 15 mM Bis–Tris, pH 7 at 4 °C) to provide improved detection of the protein bands and thence electrophoresis was performed at 300 V and 25 mA. For 15 min following the electrophoresis, the gel was incubated in reaction buffer (25 mM Tris–HCl, 5 mM MgCl2 [pH 7.4]), after which, the in-gel activity assay was performed by using a reaction mixture containing equilibrium buffer, 5 mM substrate, 0.5 mM cofactors, 0.2 mg/mL phenazine methosulfate (PMS) or dichloroindophenol (DCIP), and 0.5 μg/mL iodonitrotetrazolium (INT) in a total volume of 3 mL. For FDH-NADP, this consisted of 5 mM formate and 0.5 mM NADP whereas the FDH-NAD utilized 0.5 mM NAD instead. To confirm the presence of these enzymes the activity bands were cut and incubated in 1 mL reaction mixtures containing the corresponding substrates bicarbonate and NADPH and NADH respectively while monitoring the formation of formate via HPLCs. Fumarate reductase was detected using 5 mM fumarate and 0.5 mM formate. Confirmation was obtained by incubating the excised bands in a mixture containing fumarate (5 mM) and formate (0.5 mM) and monitoring for succinate production. Isocitrate lyase, Complex I and NADH were monitored as described in (Auger et al. 2015). Destaining solution (40 % methanol and 10 % glacial acetic acid) was used to stop the reactions where appropriate. Coomassie staining was used to ensure equal protein loading. The specificity in detections was further confirmed by performing in-gel reactions in the absence of a substrate or by the addition of the inhibitor sodium azide (5 mM). Densitometry was performed using imageJ for windows.
Spectrophotometric data for NAD-dependent isocitrate dehydrogenase (ICDH-NAD) was obtained by incubating 1 mg (protein equivalent) of membrane fraction from control and H2O2-treated cells with 2 mM isocitrate and 0.5 mM NAD for 1 min. For NADP-dependent ICDH, NADP was used instead of NAD. A similar reaction was used for malic enzyme (ME) except NAD was replaced with NADP and malate was utilized instead of ICDH. NADH and NADPH production were monitored at 340 nm over the course of a minute. Negative controls were performed without the substrates or cofactors.
Metabolite analysis
To evaluate the influence of H2O2 stress on metabolic networks, select metabolites (formate, succinate and ATP) were analysed by High Performance Liquid Chromatography (HPLC) (Alhasawi et al. 2015b). Briefly, following the harvesting of the cells at various timed intervals, the spent fluid and the soluble cellular fractions (CFE) were analyzed. An Alliance HPLC with C18 reverse-phase column (Synergi Hydro-RP; 4 μm; 250 × 4.6 mm, Phenomenex) and Waters dual absorbance detector were utilized. Mobile phase containing 20 mM KH2PO4 (pH 2.9) was used at a flow-rate of 0.2 mL/min at ambient temperature to separate the substrates and products, which were measured at 210 nm and 280 nm respectively. Peaks were quantified using the Empower software (Waters Corporation and metabolites were identified by spiking biological samples using known standards,). HPLC analyses were performed immediately after the reactions in order to minimize substrate and product degradation. Activity bands were excised from the gel and placed in 1 mL reaction mixture containing 2 mM substrates for 30 min of incubation. To monitor FDH-NAD, the excised bands were incubated in reaction mixture containing bicarbonate and NADH whereas to monitor FDH-NADP, the excised bands were incubated in reaction mixture containing bicarbonate and NADPH and in both cases, formate formation was monitored. For fumarate reductase, the reaction mixture comprised fumarate and formate while monitoring for succinate production. The sample (100 μL) was collected and diluted with 900 μL milli-Q water for HPLC analysis.
Statistical analysis
Data were expressed as means ± standard deviations. Percent change was calculated where appropriate in order to account for individual variation and to provide a better measure of the change in activity. Data were checked for significance using the student t test (p ≤ 0.05). All experiments were performed in at least biological duplicate and repeated thrice each.
Results and discussion
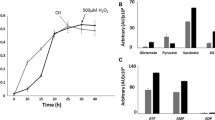
When subjected to 500 µM H2O2, P. fluorescens produced more formate in the growth media compared to the control cells at stationary phase (Fig. 1a).Footnote 1 This monocarboxylic acid is known to be a product of the detoxification of ROS by glyoxylate (Alhasawi et al. 2015b). Indeed, formate levels within the soluble cell free extract were also found to be significantly higher in the stressed cells compared to the control cells (Fig. 1b). Additionally, when whole cells were incubated with either glycine or citrate for 8 h, a sharp increase in formate levels was observed compared to the experimental controls i.e. the reaction mixtures devoid of the cells (Fig. 1c). The activity of isocitrate lyase, an enzyme known to produce glyoxylate was also found to be elevated in the stressed cultures (Fig. 1d) (Hamel et al. 2004). As formate was an important product in the cells challenged by H2O2, it was important to evaluate how this metabolite was utilized. The stressed whole cells incubated in reaction mixture containing formate consumed this carboxylic acid at a faster rate compared to the control whole cells (data not shown).

Formate production in P. fluorescens a Bacterial cell growth as measured by the Bradford assay (protein (mg/ml of culture) and formate production (monitored (by HPLC) in the growth media at various time intervals (filled square control, filled square 500 µM H2O2). b Formate levels within the soluble cell free extract (CFE). c Percent increase in formate in a whole cell experiment in which cells were taken at stationary phase from control & 500 µM H2O2 (stress) and re-suspended in control and stress media (filled square control, filled square 500 µM H2O2) containing only the carbon source (citrate) and nitrogen source (glycine) for 8 h respectively d In-gel activity of isocitrate lyase from soluble CFE obtained from control and 500 µM H2O2 culture at the same growth phase. Gels are representative of at least three independent trials. Asterisks represents statistical significance in comparison to control; n = 3, p < 0.05, mean ± SD
As formate was a key metabolite in the stressed cells, it was important to evaluate how this moiety was contributing to the anti-oxidative defence strategy of the microbe. There was a marked increase in the activity of FDH-NADP in the stressed cells (Fig. 2a).Footnote 2 In an effort to ascertain if this enzyme was being expressed as a consequence of oxidative stress, control cells were exposed to H2O2 medium while H2O2 challenged cells were incubated in a control medium. A marked reduction of FDH-NADP activity band was observed in the latter while in the former situation the activity band corresponding to the dehydrogenase was enhanced (Fig. 2b). FDH-NADP was readily inhibited by sodium azide (Fig. 2c). This enzyme may help contribute to the NADPH budget which is critical in combatting oxidative stress, as both malic enzyme and ICDH-NADP that are known to synthesize this reducing agent were also increased (Table 1) (Beriault et al. 2005, 2007; Ying 2008). The stressed cells were also characterized by an increase in FDH-NAD (Fig. 2d) which was confirmed by excision of the activity band and incubation in reaction mixture containing NADH and bicarbonate to monitor formate production (Fig. 2e). Furthermore this trend was reversed in the regulation experiment as observed (Fig. 2f). However, the presence of H2O2 resulted in the diminution of the activities of TCA cycle enzymes such as ICDH-NAD (Table 1). Also, Complex I was marked diminished in the stressed cultures (Fig. 2g) while the activity band indicative of NADH oxidase was barely evident in the control cells (Fig. 2h).

NADH homeostasis under oxidative stress. a In-gel activity of FDH-NADP. b In-gel activity of FDH-NADP following regulation experiments. c In-gel activity of FDH-NADP using azide inhibitor to ensure specificity of enzyme. d In-gel activity of NAD-dependent formate dehydrogenase. e Percent increase in formate production from incubation of excised activity bands of FDH-NAD following BN-PAGE, for 30 min in reaction mixture containing 0.5 mM NADH and 5 mM bicarbonate. f In-gel activity of FDH-NAD following regulation experiments. g In-gel activity of complex I. h In-gel activity of NADH oxidase between control and stress cells. Gels are representative of at least 3 independent trials. Asterisks represents statistical significance in comparison to control; n = 3, p < 0.05, mean ± S.D
Fumarate reductase (FRD) mediates the conversion of fumarate into succinate with concomitant oxidation of NADH (Appanna et al. 2014). Although this enzyme was present, it readily utilized formate as the reducing cofactor (Fig. 3a).Footnote 3 Formate is known to provide electrons with the liberation of CO2 (Zaunmuller et al. 2006). There was a drastic increase in formate dependent FRD in the stressed cells compared to the control cells where the activity band was only slightly visible (Fig. 3a). Regulation experiments confirmed the reversible nature of this enzyme (Fig. 3b). Furthermore, the formate dependent FRD was distinguished from the NAD-dependent FRD which also showed a marked increase in stress cells (Fig. 3c). Incubation of the excised activity band of formate dependent FRD in fumarate and formate yielded succinate (Fig. 3d). The membrane fraction from the stressed cells incubated in reaction mixture containing fumarate, formate and ADP for 30 min generated more ATP and succinate compared to the control cells (Fig. 3e).

Formate-dependent succinate production. a In-gel activity of formate-dependent fumarate reductase (FRD). b In-gel activity of formate-dependent fumarate reductase following regulation experiments. c In-gel activity of NAD-dependent fumarate reductase. d Percent increase in succinate production from excised bands following BN-PAGE incubated for 30 min in reaction mixture containing 5 mM fumarate and 0.5 mM formate. e Percent increase in metabolites (ATP and succinate) from an HPLC reaction containing Pseudomonas fluorescens membrane fraction incubated in reaction buffer with 2 mM fumarate, 0.5 mM formate and 0.5 mM ADP for 30 min (filled square control, filled square 500 µM H2O2). Gels are representative of at least 3 independent trials. Asterisks represents statistical significance in comparison to control; n = 3, p < 0.05, mean ± S.D. (0–100 %)
The data in this report point to the ability of formate to act as an important reducing factor in P. fluorescens exposed to oxidative stress. This carboxylic acid that is referred to as reduced carbon dioxide is known to provide the reducing fuel in a variety of biochemical reactions in lieu of NADH and NADPH. Reduction of ribonucleotide, nitrite and cytochrome C has been shown to be mediated by formate (Stubbe et al. 2003). In this study, formate contributes to the anti-oxidative defense strategy by supplying NADPH. Although a variety of mechanisms are deployed by microorganisms including the synthesis of exopolysaccharides (Appanna and Preston 1987) to quell oxidative tension, there is a dearth of information on such a role for this carboxylic acid. This metabolite aided by the enzymes ME and ICDH-NADP, may allow P. fluorescens to battle the oxidative challenge posed by H2O2. Indeed, various organisms are shown to evoke intricate NADPH-generating pathways to modulate their NADPH budget to combat oxidative stress (Alhasawi et al. 2014; Chenier et al. 2008; Mailloux et al. 2009). The ability of formate to reduce fumarate to succinate with the aid of FRD may prove an added benefit to this microbe as the production of NADH is markedly diminished under oxidative tension (Mailloux et al. 2011). The TCA cycle, a key generator of the catabolic reducing agent, is severely impeded (Bignucolo et al. 2013; Mailloux et al. 2007). Additionally, oxidative phosphorylation is downregulated as revealed by the diminished activity of Complex I, a situation that may impede the generation of NAD. To rectify such an occurrence, the microbe invokes the participation of NADH oxidase, an enzyme whose activity is known to be increased during environmental stress (Chenier et al. 2008). Although FDH-NAD may help alleviate the diminished NADH production in the H2O2 medium, the utilization of formate in NADH-requiring processes like in the reduction of fumarate to succinate will be an added benefit during oxidative stress. Hence, reactions necessitating NADH may switch to formate as a reducing factor. The enhanced synthesis of glyoxylate fuelled by the increased activity of ICL may argue for such a possibility. It is quite likely that P. fluorescens may have adopted this strategy. Formate, a by-product of the detoxification of ROS by glyoxylate, may have aptly been utilized by this microbe as a potent reductive power.
Although further molecular studies are required to confirm the significance of formate in anti-oxidative defence, the findings in this study argue for the possibility that Pseudomonas fluorescens may invoke the participation of this carboxylic acid in fending the challenge posed by H2O2. Formate does not only help generate NADPH but also contributes to the synthesis of key metabolites such as succinate that ensures the survival of the microbe. Hence, metabolic reconfiguration appears to be essential to the adaptation of any organism to changing environmental conditions and in this instance, an apparent by-product is retrieved to contribute to the anti-oxidative defence effort (Fig. 4).Footnote 4

Schematic demonstrating the metabolic shift involving the role of formate in P. fluorescens in combatting oxidative stress (GDH glycine dehydrogenase, GT glycine transaminase, ICL isocitrate lyase, FDH-NADP NADP-dependent formate dehydrogenase, FRD fumarate reductase-formate dependent, FDH-NAD NAD-dependent formate dehydrogenase, NO NADH oxidase, C1 Complex I). = increase in enzyme activity = decrease in enzyme activity
Notes
(These values were compared to the formate levels in the respective control experiments where the cells were omitted respectively).
These are compared to the formate values in the reaction mixture in the control and stressed bands at time 0.
These are relative to succinate levels in the control and stress reaction bands at time 0 respectively.
These values are relative to the respective metabolite levels in the reaction mixtures at time 0.
References
Alekseeva AA, Savin SS, Tishkov VI (2011) NAD-dependent Formate dehydrogenase from plants. Acta Nat 3(4):38–54
Alhasawi A, Auger C, Appanna VP, Chahma MH, Appanna VD (2014) Zinc toxicity and ATP production in Pseudomonas fluorescens. J Appl Microbiol 117:65–73
Alhasawi A, Leblanc M, Appanna ND, Auger C, Appanna VD (2015a) Aspartate metabolism and pyruvate homeostasis triggered by oxidative stress in Pseudomonas fluorescens: a functional metabolomics study. Metabolomics. doi: 10.10007s11306-015-0841-4
Alhasawi A, Castonguay Z, Appanna ND, Auger C, Appanna VD (2015b) Glycine metabolism and anti-oxidative defence mechanisms in Pseudomonas fluorescens. Microbiol Res 171:26–31
Andreadeli A, Felmatakis E, Axarli I, Dimou M, Udvardi MK, Katinakis P, Labrou NE (2009) Cloning and characterization of Lotus japonicus formate dehydrogenase: a possible correlation with hypoxia. Biochim Biophys Acta 1794:976–984
Appanna VD, Preston CM (1987) Manganese elicits the synthesis of a novel exopolysaccharide in an arctic Rhizobium. FEBS Lett 215(1):79–82
Appanna VP, Auger C, Thomas SC, Omri A (2014) Fumarate metabolism and ATP production in Pseudomonas fluorescens exposed to nitrosative stress. Antonie Van Leeuwenhoek 106(3):431–438. doi:10.1007/s10482-014-0211-7
Auger C, Appanna ND, Alhasawi A, Appanna VD (2015) Deciphering metabolic networks by blue native polyacrylamide gel electrophoresis: a functional proteomic exploration. EuPa Open Proteom 7:64–72. doi:10.1016/j.euprot.2015.05.003
Bagramyan K, Trchounian A (2003) Structural and functional features of formate hydrogen lyase, an enzyme of mixed-acid fermentation from Escherichia coli. Biochemistry 68(11):1159–1170
Bagramyan K, Galstyan A, Trchounian A (2000) Redox potential is a determinant in the Escherichia coli anaerobic fermentative growth and survival effects of impermeable oxidant. Bioelectrochemistry 51(2):151–156
Beriault R, Chenier D, Singh R, Middaugh J, Mailloux R, Appanna VD (2005) Detection and purification of glucose 6-phosphate dehydrogenase, malic enzyme, and NADP-dependent isocitrate dehydrogenase by blue native polyacrylamide gel electrophoresis. Electrophoresis 26(15):2892–2897
Beriault R, Hamel R, Chenier D, Mailloux RJ, Joly H, Appanna VD (2007) The overexpression of NADPH-producing enzymes counters the oxidative stress evoked by gallium, an iron mimetic. Biometals 20(2):165–176
Bignucolo A, Appanna VP, Thomas SC, Auger C, Han S, Omri A et al (2013) Hydrogen peroxide stress provokes a metabolic reprogramming in Pseudomonas fluorescens enhanced production of pyruvate. J Biotechnol 167:309–315
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brioukhanov AL, Netrousov AI, Eggen RIL (2006) The catalase and superoxide dismutase genes are transcriptionally up-regulated upon oxidative stress in the strictly anaerobic archaeon Methanosarcina barkeri. Microbiology 152:1671–1677. doi:10.1099/mic.0.28542-0
Chenier D, Beriault R, Mailloux R, Baguie M, Abramia G, Lemire J (2008) Involvement of fumarase C and NADH oxidase in metabolic adaptation of Pseudomonas fluorescens cells evoked by aluminum and gallium toxicity. Appl Environ Microbiol 74(13):3977–3984
Gul-Karaguler N, Sessions RB, Clarke AR, Holbrook JJ (2001) A single mutation in the NAD-specific formate dehydrogenase from Candida methylica allows the enzyme to use NADP. Biotechnol Lett 23:283–287
Hamel R, Appanna VD, Viswanatha T, Puiseux-Dao S (2004) Overexpression of isocitrate lyase is an important strategy in the survival of Pseudomonas fluorescens exposed to aluminum. Biochem Biophys Res Commun. 217:1189–1194
Hoelsch K, Suhrer I, Heusel M, Weister-Botz D (2013) Engineering of formate dehydrogenase: synergistic effect of mutations affecting cofactor specificity and chemical stability. Appl Microbiol Biotechnol 97(6):2473–2481. doi:10.1007/s00253-012-4142-9
Hourton-Cabassa C, Ambard-Bretteville F, Moreau F, Davy de Virville J, Remy R, Colas des Franc-Small C (1998) Stress induction of mitochondrial formate dehydrogenase in potato leaves. Plant Physiol 116(2):627–635
Jormakka M, Tornroth S, Byrne B, Iwata S (2002) Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295(5561):1863–1868
Jormakka M, Byrne B, Iwata S (2003) Formate dehydrogenase—a versatile enzyme in changing environments. Curr Opin Struct Biol 13(4):418–423
Lemire J, Appanna VD (2011) Aluminum toxicity and astrocyte dysfunction: a metabolic link to neurological disorders. J Inorg Biochem 105(11):1513–1517
Leonharsberger S, Korsa I, Bock A (2002) The molecular biology of formate metabolism in Enterobacteria. J Mol Micorbiol Biotechnol 4(3):269–276
Li S, Liu H, Zhang Y, Yan Y, Li Y (2009) The protective effects of α-ketoacids against oxidative stress on rat spermatozoa in vitro. Asian J Androl 12(2):247–256. doi:10.1038/aja.2009.78
Mailloux RJ, Beriault R, Lemire J, Singh R, Che´nier DR et al (2007) The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS One 2(8):e690. doi:10.1371/journal.pone.0000690
Mailloux RJ, Lemire J, Kalyuzhnyi S, Appanna VD (2008) A novel metabolic network leads to enhanced citrate biogenesis in Pseudomonas fluorescens exposed to aluminum toxicity. Extremophiles 12:451–459
Mailloux RJ, Puiseux-Dao S, Appanaa VD (2009) α-ketoglutarate abrogates the nuclear localization of HIF-1α in aluminum-exposed hepatocytes. Biochimie 91:408–415
Mailloux RJ, Lemire J, Appanna VD (2011) Metabolic networks to combat oxidative stress in Pseudomonas fluorescens. Anton Leeuwen J Microbiol 99(3):433–442
Schagger H, von Jagow G (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199(2):223–231
Shinagawa E, Toyama H, Matsushita K, Tuitemwong P, Theeragool G, Adachi O (2008) Formaldehyde elimination with formaldehyde and formate oxidase in membrane of acetic acid bacteria. J Biosci Bioeng 105(3):292–295
Singh R, Lemire J, Mailloux RJ, Appanna VD (2008) A novel strategy involved anti-oxidative defense: the conversion of NADH into NADPH by a metabolic network. PLoS One 3:2682
Stubbe J, Nocera DG, Yee CS, Chang MCY (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem Rev 103(6):2167–2202
Su C, Puls RW (2004) Nitrate reduction by zero valent iron: effects of formate, oxalate, citrate, chloride, sulfate, borate, and phosphate. Environ Sci Technol 38(9):2715–2720
Suzuki K, Itai R, Suzuki K, Nakanishi H, Nishizawa N, Yoshimura E, Mori S (1998) Formate dehydrogenase, an enzyme of anaerobic metabolism, is induced by iron deficiency in barley roots. Plant Physiol 116(2):725–732
Thomas SC, Alhasawi A, Appanna VP, Auger C, Appanna VD (2015) Brain metabolism and Alzheimer’s disease the prospect for a metabolite-based therapy. J Nutr Health Aging 19(1):58–63. doi:10.1007/s12603-014-0511-7
Uchimura H, Enjoji H, Seki T, Taguchi A, Tsakaya N, Shoun H (2002) Nitrate reductase-formate dehydrogenase couple involved in the fungal denitrification by Fusarium oxysporum. J Biochem 131(4):579–586
Yammamoto I, Saiki T, Liu SM, Ljungdahl LG (1983) Purification and properties of NADP-dependent formate dehydrogenase from Clostridium thermoaceticum a tungsten-selenium-iron protein. J Biol Chem 258(3):1826–1832
Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10:179–206
Yokota A, Kawabata A, Kitaoka S (1983) Mechanism of glyoxylate decarboxylation in the glycolate pathway in Euglena gracilis Z participation of Mn2+-dependent NADPH oxidase in chloroplasts. Plant Physiol 71:772–776
Yokota A, Koura H, Kitaoka S (1985) Refixation of photorespired CO2 during photosynthesis in Euglena gracilis Z. Agric Biol Chem 49:3309–3310
Yoshida A, Nishimura T, Kawaguchi H, Inui M, Yukawa H (2005) Enhanced hydrogen production from formic acid by formate hydrogen lyase-overexpressing Escherichia coli strains. Appl Environ Microbiol 71(11):6762–6768. doi:10.1128/AEM.71.11.6762-6768.2005
Zaunmuller T, Kelly DJ, Glockner FO, Unden G (2006) Succinate dehydrogenase functioning by a reverse redox loop mechanism and fumarate reductase in sulphate-reducing bacteria. Microbiology 152:2443–2453
Acknowledgments
This work was supported by Laurentian University and Northern Ontario Heritage Fund Corporation. Sean C. Thomas is a recipient of the Natural Sciences and Engineering Research Council Post Graduate Scholarship-Masters. Azhar Alhasawi is a recipient of a doctoral scholarship from the Ministry of Higher Education of Saudi Arabia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Thomas, S.C., Alhasawi, A., Auger, C. et al. The role of formate in combatting oxidative stress. Antonie van Leeuwenhoek 109, 263–271 (2016). https://doi.org/10.1007/s10482-015-0629-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-015-0629-6