
Abstract
Glioblastoma stem cells (GSCs) reside close to blood vessels (BVs) but vascular cues contributing to GSC stemness and the nature of GSC-BVs cross talk are not fully understood. Here, we dissected vascular cues influencing GSC gene expression and function to perfusion-based vascular cues, as well as to those requiring direct GSC-endothelial cell (EC) contacts. In light of our previous finding that perivascular tumor cells are metabolically different from tumor cells residing further downstream, cancer cells residing within a narrow, < 60 µm wide perivascular niche were isolated and confirmed to possess a superior tumor-initiation potential compared with those residing further downstream. To circumvent reliance on marker expression, perivascular GSCs were isolated from the respective locales based on their relative state of quiescence. Combined use of these procedures uncovered a large number of previously unrecognized differentially expressed GSC genes. We show that the unique metabolic milieu of the perivascular niche dominated by the highly restricted zone of mTOR activity is conducive for acquisition of GSC properties, primarily in the regulation of genes implicated in cell cycle control. A complementary role of vascular cues including those requiring direct glioma/EC contacts was revealed using glioma/EC co-cultures. Outstanding in the group of glioma cells impacted by nearby ECs were multiple genes responsible for maintaining GSCs in an undifferentiated state, a large fraction of which also relied on Notch-mediated signaling. Glioma-EC communication was found to be bidirectional, evidenced by extensive Notch-mediated EC reprogramming by contacting tumor cells, primarily metabolic EC reprogramming.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most aggressive cancer with a mean survival of less than 15 months. In spite of significant overall improvement in cancer therapy, prognosis of GBM patients remains poor and with the current best strategy of combined post-surgery radiation and temozolomide chemotherapy, survival of 10 years or more is less than 1% [1]. Addition of anti-angiogenic therapy (targeting Vascular Endothelial Growth Factor [VEGF] by Bevacizumab), while increasing progression-free survival, only marginally extends overall survival [2]. Development of better GBM therapies requires better understanding of GBM biology, particularly a better understanding how apparent intra-tumor heterogeneity affects response to therapy. A major advent in this regard was the discovery of a minor sub-population of glioma cells with stem-like properties endowed with a superior tumor-initiation capability, as well as a higher therapy refractoriness [3, 4]. Importantly, cancer stem cell (CSC) identity is not a genetically hard-wired property inasmuch as it is a context-dependent property [5]. This is reflected in the loss of CSC 'stemness' upon tumor dissociation and re-acquisition of stem-like properties by non-CSCs following re-engraftment onto a naïve recipient [6]. The contextual nature of CSC plasticity underscores a need for certain microenvironmental factors for both the induction and maintenance of CSCs stemness [7,8,9].
Among stromal elements that might play a role in the CSC niche, particular attention has been directed to blood vessels (BVs), following the pioneering study of Calabrese C et al. showing that glioma stem cells (GSCs) are not randomly dispensed but preferentially distributed in the vicinity of blood vessels [10]. Because the increase of tumor mass in fast-growing tumors usually precedes the angiogenic response, the prevailing situation in natural GBM is the co-existence of well-vascularized regions alongside under-vascularized regions experiencing a variable degree of sub-lethal hypoxia, and vast regions experiencing necrotizing lethal hypoxia. Differential availability of oxygen leads to spatially graded expression of oxygen-regulated genes, notably VEGF [11]. Non-uniform distribution of BVs similarly results in differential availability of other blood-borne substances and angiocrine factors [12, 13]. We have recently shown that non-uniform vascular distribution in cerebrally grafted GBM as well as in natural human GBM leads to a distinctive metabolic zonation driving, in turn, a non-genetic, microenvironmental phenotypic tumor cell diversification. More specifically, we showed that tumor cells within a narrow perivascular tier are distinguished from cells residing further downstream by using a non-Warburgian anabolic metabolism endowing them with more aggressive properties and a greater resistance to chemo- and radiotherapies [14]. Increased distance from perfused BVs also reduces the availability of angiocrine factors elaborated and secreted by vascular cells and shown to play multiple roles in organ homeostasis, including in the context of stem cell niches [15]. In some cases, a direct contact with tumor cells was shown to be required for transducing EC-derived signals [16]. There is some evidence that these three modes of BVs-tumor communication might also operate in the context of the GSC niche [10, 17]. Exemplifying presumed complementary contributions by the three communication modes are, respectively, regulation of the GSC marker CD133 by mTOR suggesting a metabolic regulation [18], a role for certain angiocrine factors on tumor cells invasiveness and chemoresistance properties mostly attributed to GSCs [19,20,21], and reduced CSC function in face of Notch inhibition [22, 23].
Yet, two major factors have hampered appreciating the full impact of BV-derived signals on GSCs stemness and function: first, analysis of GSC genes affected by BV-derived signals mostly relied on differential expression of GSC markers and, second, critical parameters unique to the perivascular niche (e.g., its metabolic milieu) were overlooked, as they are usually lost when pooled GSC sub-populations are used. To deal with the first problem, putative GSCs were physically isolated in a marker-independent manner, taking advantage of their relative state of quiescence. To overcome the second shortcoming, the perivascular niche and GSCs thereof were isolated under conditions preserving the authentic metabolic milieu and gene expression profile and under conditions allowing functional analyses of retrieved GSCs sub-populations.
While effects of BVs on nearby tumor cells have received considerable attention, the converse (i, e., whether and how tumor cells might affect BVs) has been largely overlooked. Of particular potential significance to tumor biology is the intriguing possibility that tumor cells might reprogram endothelial cells in ways that serve their own needs. Here, we show that GBM cells co-cultured with endothelial cells indeed re-program ECs towards increased production of key metabolites and upregulated expression of multiple cell transporters.
Materials and methods
Cell culture
Human GBM cell lines U87MG, U251MG and mouse glioma line Gl261 were obtained from ATCC and cultured in DMEM (01–055-1, Biological Industries) with 10% fetal bovine serum (FBS, 04–007-1A, Biological Industries). Human umbilical vein endothelial cells (HUVECs) tagged with RFP (Angio-Proteomie, cAP-0001RFP) were cultured in EGM™-2, endothelial cell growth medium-2 (CC-3162, Lonza) after adding all the necessary supplements provided in the kit. The cell lines were routinely examined for any possible mycoplasma contamination using Mycoplasma detection kit (LT07-118, Lonza).
Mice
Male NOD/SCID mice (8–10 weeks old) were bred and maintained at the HUJI animal facility under specific pathogen-free conditions and protocols approved by the institutional animal care committee of the Hebrew University.
Human patient samples
Patient tumor samples were surgically resected for curative reasons and were classified according to WHO classifications [24] and Grade IV GBM tumors were used for the immunostaining. All patient samples were reviewed and approved by an Institutional Review Board at Hadassah University Hospital.
Orthotopic tumor implantation
GBM cells were suspended in 1X PBS and two microliters of cell suspension (100,000 cells/µl unless indicated otherwise) were stereotactically injected after anesthetization into the striatum of the male NOD/SCID mice (8–10 weeks old) using a 10 µl Hamilton syringe [25]. Injection coordinates were 2 mm lateral to Bregma and 3 mm deep on either lobe. Injected animals were monitored for 3 weeks with the tumor burden assessed using IVIS imaging system. In all cases mice were sacrificed at the indicated time point or earlier if any signs of neurological disorder such as ataxia, paralysis, or seizures were observed.
Sorting cancer cells based on their relative distances from perfused BVs
Tumor-bearing mice were injected with a vascular perfusion dye (Hoechst 33,342 (Sigma), 5 mg/kg body wt) through the tail vein followed by mice euthanization and sacrifice within five minutes from dye infusion. The brain was resected, the tumor separated under a fluorescent stereomicroscope (Nikon), and tumor cells were dissociated using tumor dissociation kit (130–096-730, MACS Miltenyi Biotec). Single-cell suspensions were FACS-sorted (with the gating adjusted for exclusion of cell doublets and all stromal cells) according to their level of Hoechst 33,342 dye uptake on the premise that tumor cells with the highest level of dye uptake are perivascular tumor cells. A detailed protocol of this procedure was published in Bio-protocol [25].
Neurosphere assay
Cultured glioma cells (U87MG or Gl261) or pre-sorted cells from U87MG-GFP xenografts were plated onto a non-adhesive dish at a low seeding density of 1000 cells per 10-cm culture plate and grown in neurosphere assay growth medium containing serum-free DMEM supplemented with B27 Supplement (17,504,044, ThermoFisher Scientific), epidermal growth factor (236EG, R&D Systems, 20 ng/ml), and basic fibroblast growth factor (233FB, R&D Systems, 10 ng/ml). Media was replaced every 72 h and neurosphere formation was inspected under a fluorescent microscope (Nikon, USA).
Limited dilution assay for tumor-initiation potential
Serial tenfold dilutions of cancer cells (at the range of 105–102 cells per mouse) were transplanted into the striatum of the NOD/SCID mice and appearance of tumors was monitored as described above. Limiting dilution analysis was performed using ELDA online tool (https://bioinf.wehi.edu.au/software/elda/) [26]. Cryosections including the injection site were examined for validating cases of no tumor development.
Analysis and isolation of label-retaining tumor cells
The indicated tumor cells were stained with PKH26 dye (PKH26GL, Sigma) for 2 min (1 µl for 0.1 million cells in 1 X PBS), blocked and washed with DMEM containing 10% FBS. For ex-vivo analysis, pre-labeled cells were plated at a low density under the conditions used for neurosphere formation. For in vivo experiments, PKH26 stained cells were immediately inoculated intracranially into the striatum of the mice as described above. Label-retaining cells in tumor cryosections were visualized by direct PKH26 fluorescence. PKH26 fluorescence was also used for sorting label-retaining cells (presumed relatively quiescent tumor cells) from single-cell tumor suspensions or, specifically, from the perivascular niche. For the latter, tumor cell population positive for both Hoechst 33,342 and PKH26 were sorted.
Flow cytometry
Cells were trypsinized or tumors were dissociated and single-cell suspension of 106 cells were either stained for specific antibodies against CD133-PE-Vio770 (1:50, 130–111-081, MACS Miltenyi Biotec), CD271-APC (1:50, 130–112-602, MACS Miltenyi Biotec) for 20 min, washed and analyzed in FACS buffer using Aria III or Aria LSRFortessa flow cytometer (BD Biosciences). Results were analyzed using Flow Jo software after appropriate gating. Cells were gated using forward scatter (FSC) and side scatter (SSC) (A stands for Area). Doublet discrimination was performed to exclude doublets from the study by choosing cells within the Width (W) and Height (H) parameters of SSC followed by FSC. Stromal exclusion was done by filtering out all the non-GFP labeled cells (U87MG-GFP gate). Fractionation of cells according to their relative distance from blood vessels was performed by sorting cells based on their Hoechst intensity (H + vs H-) and the label retaining (putative GSCs) were isolated based on their retention of PKH26 dye (P + vs P-). H + P + U87MG-GFP cells were dubbed as perivascular GSCs.
Cryosectioning
Tissues were fixed in freshly prepared 4% paraformaldehyde (PFA, 15,710, Electron Microscopy Sciences) for 4–6 h at 4 °C, transferred to 30% sucrose solution and kept for 18 h at 4 °C. Tissues were then embedded in OCT (4586, Scigen) and 10–50-µm-thick sections were cut in the cryostat at—20º C and collected onto a poly-L-lysine-coated glass slides.
Immunostaining
Tissue sections were rehydrated in 1X PBS, permeabilized and blocked in 3% bovine serum albumin (BSA, 0332-TAM-50G, VWR Chemicals) containing 1X PBS with 0.01% Tween-20 (PBST) for 2 h at room temperature followed by overnight incubation with CD31 antibody (1:100, 550,274, BD Pharmingen) at 4 °C. For human patient samples, cryofrozen tissues were sectioned and fixed with 4% PFA for 5 min and the above-mentioned immunostaining protocol was repeated with human-specific antibodies against CD271 (1:100, 345,101, Cell Signaling), CD31 (1:100, ab28364, Abcam). Slides were washed thrice in 1X PBST, incubated with a secondary antibody for 2 h at room temperature, washed thrice with 1XPBST and mounted. Secondary antibodies used were Cy5-IgG-Rat (1:500, 112–175-143) and Cy3-IgG-Rabbit (1:500, 111–165-003) both from Jackson Immunoresearch Laboratories Inc.
Knockdown and stable cell line generation
Lentiviral vectors containing shRNA against human CD271 (HSH102902, GeneCopoeia) and shRNA against LIF were purchased from Santa Cruz Biotechnology Inc. 15 µg of the respective vector, 12 µg of transfer plasmid (pMD2.G), and 3 µg of packaging plasmid (psPAX2) were co-transfected into HEK 293 T cells. Supernatant containing lentivirus particles were harvested 48–72 h post infection, filtered and used to infect U87MG or U251MG cells in the presence of 6 µg/ml of polybrene. Infected cells were expanded to 10 cm dishes from a 6-well plate 48–72 h post infection, and sorted for mCherry-positive clones using BDFACS Aria III sorter. Knockdown efficiency in stable lines was evaluated by RT-PCR.
Co-culture
HUVEC-RFP (HR) cells and Gl261-GFP (GG) cells were plated together overnight in EGM-2 medium in T75 flask at a seeding density ranging from 100,000 to 500,000 cells such that they reach a cell ratio of − 3:1 or − 1:3 (HR: GG) 72 h later. Cells were then trypsinized and suspended in fresh EGM-2 medium. A small fraction of it was analyzed by FACS to determine the final HR:GG ratio. For comparison with separately cultured HR and GG cells, the two cell lines were grown separately in EGM-2 medium and mixed at the exact same ratio (named Sep mixture). Samples were quickly lysed and RNAs were subjected to RNA-seq analysis. For Notch inhibition, Gamma secretase inhibitor (GSI) (SCP0004, Sigma) was added to the co-culture or to the separately cultured cells at 20 µM for 24 h before harvesting.
RNA Extraction and quantitative RT-PCR
RNAs were extracted using Trizol reagent (T9424, Sigma) and cDNAs were prepared using RNeasy mini kit (74,104, Qiagen). Quantitative real-time RT-PCR was performed using SYBR-Green PCR Master mix (A46109, ThermoFisher Scientific). qPCR primers used in this study are listed in Supplementary Table 1.
Transcriptomics
Quality of RNAs prepared from sorted fractions was examined with the aid of a TapeStation (Agilent Technologies). Libraries were prepared and sequenced using Illumina directional RNA sequencing protocol (Hi-Seq). Reads were mapped using TopHat2, quantified and normalized using Cuffdiff to produce gene level-normalized expression values [27]. Differentially expressed genes were defined as differences with a p-value less than 0.05. For mTOR inhibition studies, total RNA was isolated from – 50,000 cells and CEL-Seq2 analysis was performed [28].
For unambiguous identification of RNAs as human or mouse transcripts in co-culture experiments and to avoid interference due to ambiguity with the differential gene expression analysis, both alignment files (the results of mapping to both human and mouse) were used to determine the true origin of each read. RNA-Seq was performed to obtain longer reads (100 bp) to increase the efficiency of unambiguous alignment. An in-house Perl script compared the quality of mapping, essentially by alignment scores, and separated the reads into human origin, mouse origin, or undetermined (where the read mapped equally well to both genomes, which was less than 1% of the total reads and were discarded). Sequences were mapped to the genome version GRCh38 using the respective species-specific annotation from Ensembl release 78. Reads were then mapped using TopHat2 and normalization was done using Cuffdiff as explained above. Identification of biological pathways enriched in different datasets was determined by GSEA analysis. Heatmaps were constructed using R program for the top differential genes in the particular dataset. Metabolic gene expression analysis and exploratory analysis of co-cultured RNA-seq data were performed using a BIOMEX analysis platform [29]. All RNA Sequencing data are available in the ArrayExpress database under accession number: E-MTAB-9610, E-MTAB-9607, E-MTAB-9604, and E-MTAB-6882.
IVIS imaging
For monitoring tumor growth in living animals, U87MG and U251MG cells were transduced with mCherry by lentivirus infection and established cell lines were implanted intracranially into the NOD/SCID male mice as explained earlier. Mice were imaged for mCherry fluorescence intensity using an IVIS imaging system (PerkinElmer).
Statistical analysis
Descriptive statistics were generated for all the quantitative data with presentation of means ± SEM. Significance was tested by either t-test or ANOVA and represented using Graphpad Prism software.
Results
Marker-independent isolation and characterization of perivascular GSCs
To isolate the perivascular niche from GBM tumors, green fluorescent protein (GFP)-tagged U87MG tumor cells were intra-cerebrally grafted and 3 weeks later mice-bearing U87MG-GFP tumors were intravenously infused with a fluorescently labeled, freely diffusing perfusion dye (Hoechst 33,342). To allow limited diffusion, tumors were retrieved within 5 min from dye infusion. Tumors were then dissociated and stroma-free viable GFP-tagged tumor cells were sorted on the basis of Hoechst dye uptake on the premise that dye uptake will be mostly confined to tumor cells closest to perfused BVs (Fig. 1A). Cells derived from successive sorted fractions representing progressive distances from the nearest perfused BVs (dubbed as Hh—Hoechst high, Hm—Hoechst medium and Hl—Hoechst low) were collected and analyzed with respect to GSC distribution, gene expression patterns, and biological traits. The three successive sorted fractions had no significant difference in their cell viability (Supplementary Fig. 1A). The proof for the claim that this methodology helps us to isolate cancer cells with differ significantly in their perfusion limits was shown by analyzing the hypoxia-related genes in two sub-fractions at the opposite end of the perfusion gradient spectrum (perivascular Hh and hypoxic Hl). The three hallmark genes upregulated during hypoxia namely—Glucose Transporter 1 (GLUT1), Carbonic Anhydrase 9 (CA9), and Phosphoglycerate Kinase 1 (PGK1) were significantly upregulated in Hl fraction indicating the relative limited diffusion of oxygen in these regions (Supplementary Fig. 1B). In a previous study, we have also shown that Hh fraction is composed of tumor cells residing within 60 μm from perfused vessels, dubbed the 'perivascular niche' that enjoys greater perfusion [14].

GSCs are clustered in a perivascular niche. A A procedure for tumor cell fractionation based on their relative distance from BVs (see Methods, Abbreviations: SSC side scatter, FSC forward scatter, A area, W width, and H height). B Neurospheres generated by U87MG-GFP cells residing at increasing distance from perfused BVs (cell fractions dubbed Hh, Hm, and Hl, with the former representing tumor cells closest to BVs). C Quantification of the number (left panel) and size (right panel) of neurospheres. n = 5, Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01. D Tumor-initiation capacity of the U87MG-GFP cells retrieved from the respective three fractions and transplanted into the striatum of NOD/SCID mice. Last column signifies the minimum no. of cells required for tumorigenesis in respective fractions
To provide a functional evidence that GSCs are indeed clustered in the perivascular niche, we first examined their neurosphere-generating capacity, a commonly used surrogate assay for cerebral GSC activity. As shown in Fig. 1B, C, Cells within the Hh tier formed significantly more neurospheres, as well as neurospheres of a larger size compared to more distantly located tumor cells. Analysis of tumor-initiation potential, the gold standard assay for GSC activity, corroborated that cells within the perivascular tier are endowed with a higher tumor-initiation activity compared to that of cells residing further downstream (Fig. 1D), thus providing a marker-independent, functional confirmation that GSCs are preferentially clustered in the perivascular niche.
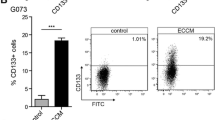
Because the utility of surface makers expression as an unambiguous, general CSC identifier has been challenged [30], we carried out an unbiased transcriptional analysis of tumor cells retrieved from the perivascular niche and sub-selected based on a biological trait distinguishing GSCs from non-GSCs residing in the same microenvironment. We exploited the ‘quiescence’ property of GSCs relative to other rapidly dividing cancer cells and thus employed a label-retention strategy to identify putative CSCs using a membrane-associated dye - PKH26, that selectively labels slow-dividing cancer cell population [31]. In preparatory ex-vivo experiments, U87MG-GFP cells uniformly pre-labeled with fluorescent PKH26 were cultured and shown that progressive dilution of PKH26 can indeed serve as a reliable cell division counter (Supplementary Fig. 1B). Next, PKH26-tagged U87MG-GFP cells were seeded in a neurosphere enrichment medium and grown in 3D configuration followed by sorting dissociated cells into PKH26-high and PKH26-low fractions (dubbed as P + and P- cells, respectively). Results validated a co-segregation of PKH26-retaining cells with CD133, an established GSC marker (Supplementary Fig. 1C, D). More importantly, PKH26-retaining cells were shown to have superior tumor-initiation capabilities compared to label non-retainer cells (Supplementary Fig. 1E).
For selective labeling of the perivascular niche in vivo, U87MG-GFP cells pre-labeled with PKH26 were orthotopically implanted into 8-week-old male NOD-SCID mice and tumors were allowed to grow for 3 weeks prior to i.v. infusion of Hoechst 33,342 and tumor retrieval 5 min thereafter. Co-visualization of Hoechst 33,342-positive cells and PKH26-retaining cells in tumor sections showed that the latter are mostly localized in the perfusion-rich region of < 60 μm from BVs (Fig. 2A, and Supplementary Video 1). Combinatorial FACS analysis of H + cells and P + cells corroborated profound enrichment of label-retaining cells in the perivascular tier (Fig. 2B, C). Quantitatively, approximately 5% of H + cells were also P + (Fig. 2D), suggesting that this is also the estimated GSC frequency in the perivascular niche. Other than providing an additional functional proof for GSCs clustering in the perivascular niche, these results have provided us with a way for marker-independent elucidation of transcriptional signatures of perivascular GSCs. To this end, isolated GCSs (the H + P + population) were subjected to RNA-seq analysis alongside non-GSC cells from the same microenvironment (the H + P- population). Reinforcing the notion that H + P + cells represent perivascular GSCs, H + P + cells showed upregulated expression of a large number of genes previously recognized as GSC markers, as well as of CSC markers described for other tumors but not for glioblastoma (see Supplementary Fig. 2A, B for a heat map and for validation/qRT-PCR quantification of representative GSC markers and other stem cell-related genes). A notable example for the latter group is CD271 (also referred to as nerve growth factor receptor (NGFR), highlighted by arrow in Supplementary Fig. 2A) previously implicated in multiple cancers such as melanoma, head and neck cancer [32,33,34]. More importantly, a third category of uncovered differentially expressed GSC genes included a large group of genes not previously recognized as CSC marker genes in either glioblastoma or in other tumors (Fig. 2E).

Marker-independent isolation and transcriptomic profiling of perivascular GSCs. A Above panel A representative confocal image of a grafted U87MG-GFP tumor section highlighting clustering of label-retaining PKH26 + tumor cells (pseudo-colored white) in the perfusion-rich perivascular niche. Below panel quantification of PKH26 + tumor cells in the indicated cellular tiers representing increased distances from perfused capillaries. B Combined fractionation and FACS gating for PKH26-positive cells (dubbed P +) and Hoechst 33,342-positive cells (dubbed H +) from intracranially implanted U87MG-GFP tumors, where H + P + cells considered as perivascular GSCs and H + P- cells as non-GSC perivascular cancer cells. (n = 10) C FACS distribution and relative quantification of mean fluorescence intensity in (MFI) of sorted cell populations showing enrichment of label-retaining cells (P +) in the perivascular region. (n = 5) D Quantification of P + cells (as % of total) in perfused (H +) or non-perfused (H-) tumor regions. (n = 5) E Heatmap showing top 50 differentially expressed genes distinguishing perivascular GSCs (H + P +) from non-label-retaining cells (H + P-). Shown are data from two biological repeats and their average. F GSEA plots showing the top 4 pathways enriched in the H + P + fraction. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001
Given that perivascular GSCs were isolated based on their relative quiescence, it was anticipated, and indeed found that differentially expressed genes in H + P + cells included multiple genes implicated in cell cycle regulation. Outstanding among the top 50 upregulated H + P + genes were genes involved in cell cycle arrest, exemplified by the P53 target genes CDKNIA and CDKN2D and additional genes implicated in the G2M checkpoint [35,36,37] (Fig. 2E). Conversely, downregulated in H + P + cells included genes known to enhance cell cycle progression, exemplified by CDK15 and CDK18. GSEA analysis highlighted the G2M checkpoint regulator genes, P53 pathway, ABC transporters, and mTORC1 signaling as additional pathways enriched in perivascular GSCs (Fig. 2F).
The potential contribution of the newly identified marker genes to stem cell identity and tumorigenesis is yet to be determined. Here, we only examined the functional significance of upregulated CD271 expression, prompted by the findings that it is mostly expressed in H + P + cells (Fig. 2E, Supplementary Fig. 3A and 3B), whereas its ligand, NGF, is elaborated by nearby endothelial cells (EC expression data to be shown below). To this end, a CD271 knockdown strategy was used with lentivirus-mediated ex-vivo transduction of CD271-specific shRNAs in a vector also containing a ubiquitously expressed mCherry reporter to aid monitoring tumor growth following orthotopic implantation in NOD-SCID mice (for the efficiency of CD271 knockdown see Supplementary Fig. 3C). Even partial knockdown of CD271 led to significant reduction of tumor-initiation capability and reduction of tumor size (see Supplementary Fig. 3D-F for grafted U87MG cells and Supplementary Fig. 3G-I for grafted U251MG cells). U87MG xenografts were further analyzed for their distribution of micro-vessels by staining with CD31 (BV marker). Micro-vessel density (MVD) quantification also revealed a decreased vascular coverage in tumors expressing low levels of CD271 compared to the control (Supplementary Fig. 3L and 3 M). These results indicate that the previously documented contribution of CD271 to glioma tumorigenesis and the correlation between CD271 expression and glioma prognosis [38, 39] are mostly attributed to its production by perivascular GSCs.
The metabolic milieu of the perivascular niche shapes the GSC transcriptome
In light of our previous study showing that metabolism of perivascular tumor cells differ from the metabolism of tumor cells located elsewhere [14], we examined whether and how the unique metabolic milieu of the perivascular niche contributes to the properties of perivascular GSCs. Given the pivotal role of mTOR as integrator of multiple environmental inputs together with our finding that mTOR activity in glioblastoma is mostly confined to a narrow perivascular tier [14], we examined the effect of mTOR inhibition on perivascular GSCs gene expression. It should be pointed out that mTOR inhibition was previously shown to compromise tumor-initiation capability of glioblastoma-derived cells [40,41,42], however, ascribing reduced tumorigenicity to reduced activity of a particular mTOR-controlled GSC marker gene(s) remains to be demonstrated. To this end, U87MG-GFP tumor-bearing mice were treated with the mTOR inhibitor Torkinib that targets both mTOR complexes for 6 consecutive days prior to tumor retrieval. Perivascular niches from Torkinib-treated tumors (or from tumors treated with vehicle only) were then isolated and subjected to a comparative RNA-seq analysis. As seen in the heat maps presented in Fig. 3A, mTOR inhibition using Torkinib (inhibitor of both mTORC1 and mTORC2 complexes) had a major impact on the expression of GSC signature genes, both on previously recognized CSC markers, as well as on GSC markers uncovered by this study. Exemplifying the latter group are the two P53 target genes, the ABCC3 transporter and the Wnt receptor FZD8 previously shown to play a role in drug resistance [43,44,45] and shown here to be preferentially expressed in perivascular GSCs in an mTOR-dependent manner.

mTOR-regulated expression and activity of perivascular GSCs A Heat maps of RNA-seq data highlighting mRNAs in the perivascular niche of U87MG tumors downregulated upon mTOR inhibition (Veh vehicle, Tor torkinib). Left transcripts of known GSC marker genes. Right transcripts of GSC marker genes uncovered by this study. B Preferential expression of CD271 in perivascular (Hh) vs. distal (Hl) U87MG-GFP cells expressed as the number of fragments per KB of exon per million reads (FPKMs) (n = 2) on left and validated by q RT-PCR (N = 3) on right. C Downregulation by Torkinib of CD271 expression in the perivascular niche of U87MG-GFP tumors (n = 3) expressed as in B. Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01. D Box-whisker plot showing the expression of CD271 in normal and GBM patient samples obtained from TCGA and analyzed using web portal UALCAN [61]. E Kaplan–Meier plot showing the association of CD271 expression levels with patient survival. F Immunostaining of untreated primary GBM human patient sample showing elevated expression of CD271 (green) near the tumor vasculature (CD31: red). G Quantification of CD271 in GBM patient samples as a function of distance from nearest capillaries. (n = 5) Data represented as mean ± SEM. **p < 0.01
Likewise, expression of the GSC marker gene CD271 by perivascular GSCs was also found to be mTOR-dependent (Fig. 3B, C), arguing for a metabolic beneficence to its apparent major contribution to tumor initiation (Supplementary Fig. 3 J, K). CD271 expression was also found to be elevated in human GBM patients (Fig. 3D) and more importantly, we found that elevated expression of CD271 is correlated with worse GBM patient survival (Fig. 3E). Taken together, these results show that apparent clustering of CSCs near perfused BVs is, at least in part, due to the supportive metabolic milieu of the perivascular niche.
Perfusion-independent and contact-dependent vascular cues contributing to GSC stemness
Increased awareness for the role of angiocrine factors in cancer stem cell biology, in general, has suggested a role for angiocrine factors in the maintenance of GSC stemness. Perfusion-independent contributions of ECs to tumor initiation and propagation were indeed reported by several groups based on tumor-ECs co-culture systems [12, 13, 22, 46, 47]. With the aims of further characterizing perfusion-independent vascular influences, as well as for identifying pathways relying on direct BVs-tumor cell contacts, we carried out the following experiments: ECs from human umbilical veins tagged with RFP (HUVEC-RFP) were co-cultured with mouse glioma line Gl261 fluorescently tagged with GFP (Gl261-GFP). Upon reaching confluency by 72 h post seeding, cells were harvested and RNAs extracted. The combined co-culture transcriptomes were determined from generated cDNA libraries without prior separation of the respective cell types (sample named as Co), taking advantage on the fact that ECs and GBM cells were of different species thus allowing distinguishing GBM (mouse) and EC (human) transcripts. A comparison was then made with the EC and GBM transcriptomes elucidated for an artificial mixture of the two cell types (Gl261-GFP and HUVEC-RFP) retrospectively prepared at the exact ratio of cells reached by the end of the co-culturing (as determined by FACS-based quantification of the GFP/RFP ratio) (sample named Sep) (See Fig. 4A for a scheme and 'Methods' for further details). Considering that contact-dependent EC signaling is often regulated by canonical Notch signaling [48] and previous findings implicating Notch signaling in GSCs self-renewal [47, 49], GBM–EC co-cultures were also treated with the Notch pathway inhibitor Gamma secretase inhibitor (GSI) (or with the DMSO vehicle alone) for 24 h before harvesting.

Perfusion-independent vascular contributions to glioma stemness revealed by EC-GBM co-culturing. A Co-culture setup for identifying perfusion-independent vascular cues affecting GSCs properties. B Principal component analysis (PCA) of separately (Sep) or co-cultured (Co) cells without (Veh) or with the Notch inhibitor GSI. C Heat map showing differentially regulated glioma genes in the four different experimental settings specified above. Enclosed in dashed boxes are the following group of genes: pro-differentiation genes downregulated in the presence of ECs (green), anti-differentiation genes upregulated in the presence of ECs in a Notch-dependent manner (blue), anti-differentiation genes upregulated in the presence of ECs in a Notch-independent manner (red). Highlighted by arrows are the anti-differentiation genes LIF and Id1 further analyzed in D and E. (n = 3) D Mouse glioma cell line (Gl261-GFP:GG) and human endothelial cell line (HUVEC-RFP:ER) were seeded at the indicated cell ratios and harvested upon reaching confluence (top image). LIF and Id1 transcripts in glioma cells were determined by quantitative RT-PCR using mouse-specific primers normalized to mouse beta-actin (n = 3). Data are presented as mean ± SEM. *p < 0.05 and **p < 0.01. E Tumor-initiation assay of U251MG-mCherry line expressing control shRNA or LIF-specific shRNAs
Principal component analysis (PCA) of GBM transcripts comparing Co and Sep revealed a clear divergence indicative of significant modulation of the GBM transcriptome by EC-derived signals (Fig. 4B, left panel). Likewise, there was a clear divergence between co-cultures treated, or not, with GSI (dubbed as Co_GSI and Co_Veh, respectively), indicating a role for Notch-mediated, contact-dependent GBM–EC communication. Noteworthy, in the absence of ECs, Notch inhibition had little effect on the GBM transcriptome (Sep_GSI vs. Sep_Veh), highlighting a role for Notch signaling in heterotypic GBM–EC communication but not for homotypic GBM cells interaction (Fig. 4B, right panel).
Outstanding among the many GBM genes affected by neighboring ECs were multiple genes implicated in differentiation control, manifested by upregulated expression of more than a dozen genes known to possess a net anti-differentiation activity and, conversely, by down-regulated expression of genes reported to drive cell differentiation (e.g., Myc, Kitl, Bdnf) (within green box of Fig. 4C). Remarkably, a significant fraction of differentiation-related genes (but not all of them) relied on direct GBM–EC cell contacts, evidenced by the finding that GSI treatment brought-down their expression to a level comparable to that detected in separately cultured GBM cells (Fig. 4C). Exemplifying anti-differentiation genes whose expression in GBM is upregulated by EC-derived signals are ID1 and LIF representing, respectively, Notch-independent and Notch-dependent expressions (blue and red arrow in Fig. 4C respectively). As anticipated, increasing the EC-to-GBM ratio led to a further increase of both ID1 and LIF expressions in GBM cells (use of species-specific PCR primers allowed quantifying the respective RNAs exclusively in GBM cells), presumably via the increase in GMB-EC contacts (in the case of LIF) and/or via increased availability of angiocrine factors (in the case of ID1) (Fig. 4D).
To provide a functional proof that LIF contributes to GSC stemness, we examined the consequences of LIF knockdown on tumor-initiation potential as a readout for GSC activity. To this end, U251MG cells expressing pre-selected shRNAs against LIF (Supplementary Fig. 4A–C) were grafted in the brain of naïve mice. Results showed that LIF knockdown significantly reduced tumor-initiation capability relative to U251MG cells transduced with vector only (Fig. 4E). Taken together, these results suggest a key role of perfusion-independent EC-derived signals in the prevention of GSC differentiation.
Another notable pathway enriched in GBM owing to EC presence was epithelial mesenchymal transition (EMT) (Supplementary Fig. 4D). Considering previous studies showing that EMT conditions tumor cells acquire stem cell properties [50, 51], this finding points at another possible way by which proximity to BVs may promote GSC stemness. The finding that Notch pathway inhibition lowers EMT enrichment score (Supplementary Fig. 4D) suggests that glioma cells contacting ECs are more EMT prone.
Transcriptional reprogramming of ECs by adjacent/contacting glioma cells
The glioma-EC co-culture provided us with an opportunity, not only to reveal GBM genes affected by EC-derived signals but also for the converse, i.e., to reveal EC genes affected by GBM-derived signals. To this end, the same RNA preparations obtained from GBM:EC co-cultures (or from separately cultured cells) and used above for analysis of GBM-derived transcripts were also analyzed for EC-derived transcripts, with the latter identified on the basis of their human origin. Compatible with the proposition of glioma-instructed EC 'education,' PCA analysis showed a clear divergence between the transcriptomes of ECs cultured alone or together with GBM cells (Fig. 5A). As done vis-à-vis GBM transcripts, inclusion of a Notch pathway inhibitor in the culture medium suggested that EC reprogramming by GBM signals is, to a large extent Notch-dependent.

Metabolic reprogramming of ECs by Glioma. A PCA analysis of EC transcripts in the four experimental setting used for analysis of glioma transcripts (named as specified in the legend of Fig. 4B). B GSEA of the EC transcriptome with the top panel highlighting the indicated pathways enriched by co-cultured glioma cells and bottom panels showing their level of dependence on Notch signaling. C Glioma-induced EC reprogramming deduced from EC transcriptomic data with the aid of a BIOMEX analysis platform developed in the Carmeliet laboratory [29]. Green arrows indicate metabolic pathway upregulated in the presence of glioma cells (Co vs Sep). Note nullification of the metabolic effects exerted by glioma cells in the presence of GSI (Co GSI vs Co). D Heat map highlighting EC genes affected by co-cultured glioma cells in a Notch-dependent manner. Exemplifying metabolic genes are genes involved in serine metabolism (red arrows). Also highlighted are affected amino acid transporters (blue arrows). E Heat map of differentially expressed endothelial (left panel) and glioma (right panel) genes encoding solute carrier transporters. n = 3 in all experiments
GSEA analysis of endothelial transcriptome highlighted metabolic reprogramming of ECs as a process markedly impacted by glioma cells inputs, including, alterations in protein secretion, amino-acyl tRNA biosynthesis, and amino acid metabolism (Fig. 5B). Comparative analysis of the 1,511 metabolic genes expressed in our dataset (see 'Methods' for details) showed that metabolic reprogramming of ECs encompasses almost all major metabolic pathways and, moreover, that nearly all glioma-induced EC changes are nullified in the face of Notch inhibition (Fig. 5C). Notch dependence of glioma-instructed EC alterations was also evidenced from the heat map highlighting the top 50 affected EC genes which, in addition to key metabolic genes (e.g., PHGDH, PSAT1 engaged in serine biosynthesis), also included multiple solute carriers (e.g., SLC7A11, SLC1A4 engaged in the transport of amino acids) (Fig. 5D).
Discussion
Findings that CSC identity is context-dependent and that CSCs reside in close proximity to BVs brought up the question what makes the perivascular microenvironment unique in this respect. To address this issue, we harnessed our newly developed methodology devised for isolation of perivascular tumor cells under conditions allowing a direct comparison between perivascular niche-resident GSCs and GSCs located further away from perfused BVs. Importantly, GSCs were isolated from the respective microenvironmental tiers not on the basis of a particular marker expression but rather on a functional basis, exploiting their relative quiescence compared to non-GSCs from the same microenvironment. A marker-independent GSC isolation was used to extend the list of GSC signature genes beyond the list of currently recognized CSC markers. A notable group of differentially expressed GSC genes uncovered by this study were multiple genes implicated in cell cycle control and likely responsible for GSC quiescence.
The presence of vast under-vascularized regions in GBM renders adjacency to BVs a major determinant responsible for limited availability of oxygen and other blood-borne substances and a resultant metabolic zonation. Not surprisingly, therefore, our results showed that the unique metabolic milieu of the perivascular niche, and, specifically, the restricted zone of mTOR activity confined to this niche is essential for the maintenance of GSC stemness and can explain why GSCs are clustered within < 60 μm from the nearest perfused BV. Yet, our data show that not more than 5% of tumor cells in this microenvironment acquire and/or maintain GSC properties. The question why the majority of tumor cells exposed to identical vascular inputs are not GSCs remains enigmatic (lateral inhibition?). Inhibition of notch signaling as a therapeutic approach has unfortunately rendered limited success and has not passed beyond clinical trials due to gastrointestinal toxicity issues [52]. GSIs, even though have been very efficient in sensitizing cell lines to radiation and targets GSCs both in vitro and in vivo [53, 54], therapeutic resistance to these notch inhibitors has been observed [55]. This is partly due to the activation of alternative pathways through CDK9-mediated transcriptional elongation or over-activation of Hedgehog pathway [56, 57]. Thus, the need of the hour is the combinatorial therapy of notch inhibition along with alternative pathways with limited toxicity and greater specificity.
While conducive to stemness, differential availability of blood-borne substances cannot on its own account for the full spectrum of GSC properties and must be complemented by the action of angiocrine factors with a limited diffusion range. The GBM-BV co-culture system analyzed herein indeed uncovered a non-overlapping set of genes impacted by the presence of BVs. Remarkably, it appears that activities responsible for maintaining GSCs in an undifferentiated state are mostly mediated by perfusion-independent factors. A surprisingly large fraction of perfusion-independent vascular signals was shown to rely on direct glioma-BV heterotypic cell contacts, evidenced by their nullification by Notch pathway inhibition. Notch signaling was shown to be elevated in perivascular GSCs in a study by Bayin et al. [58]. In addition, they observed that Notch signaling regulates metabolic adaptations to the microenvironment. Our study is also in alignment with their findings exemplified by changes in the GSC signature upon treatment with major metabolic inhibitor Torkinib (Fig. 3A). Taken together, three different modes of tumor-BV communication, namely signals mediated by blood-borne substances, angiocrine signals, and Notch-mediated direct cell-to-cell signaling appear to play complementary roles in GSC biology.
The finding that at their interface tumor-BV communication is bidirectional, i.e., that tumor cells also reprogram endothelial cells is of much interest. Of particular significance is our finding that tumor cells enhance multiple biosynthetic metabolic pathways in adjacent ECs in a Notch-dependent manner. This brings up the intriguing possibility that tumor cells reprogram endothelial cells for the purpose of providing them with needed metabolites. The finding of reciprocal upregulated expression of multiple transporters on both ECs and tumor cells (Fig. 5E) is compatible with this rather speculative proposition. A two-sided upregulation of multiple cellular transporters raises the intriguing possibility of metabolites exchange between tumor cells and BVs.
One of the key challenges that need to be addressed in the near future should include the use of brain-specific primary ECs to study the brain tissue-specific and species-specific interactions that we might have missed due to our cross-species in vitro cell culture system using HUVECs [59, 60]. Perfusion-dependent contribution of ECs can be further delineated from contact-dependent influence using trans-well barriers between glioma and ECs. Taken together, these findings underscore multiple levels of tumor cells-BVs interplay, and delineate different mechanisms by which proximity to BVs secures GSC stemness.
References
Tykocki T, Eltayeb M (2018) Ten-year survival in glioblastoma A systematic review. J Clin Neurosci 54:7–13. https://doi.org/10.1016/j.jocn.2018.05.002
Batchelor TT, Reardon DA, de Groot JF, Wick W, Weller M (2014) Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res 20(22):5612–5619. https://doi.org/10.1158/1078-0432.CCR-14-0834
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN (2015) Cancer stem cells in glioblastoma. Genes Dev 29(12):1203–1217. https://doi.org/10.1101/gad.261982.115
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432(7015):396–401. https://doi.org/10.1038/nature03128
Meacham CE, Morrison SJ (2013) Tumour heterogeneity and cancer cell plasticity. Nature 501(7467):328–337. https://doi.org/10.1038/nature12624
Magee JA, Piskounova E, Morrison SJ (2012) Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21(3):283–296. https://doi.org/10.1016/j.ccr.2012.03.003
Thankamony AP, Saxena K, Murali R, Jolly MK, Nair R (2020) Cancer stem cell plasticity—a deadly deal. Front Mol Biosci 7:79. https://doi.org/10.3389/fmolb.2020.00079
Poli V, Fagnocchi L, Zippo A (2018) Tumorigenic cell reprogramming and cancer plasticity: interplay between signaling, microenvironment, and epigenetics. Stem Cells Int 2018:4598195. https://doi.org/10.1155/2018/4598195
Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S, Brons NHC, Leite S, Sauvageot N, Sarkisjan D, Seyfrid M, Fritah S, Stieber D, Michelucci A, Hertel F, Herold-Mende C, Azuaje F, Skupin A, Bjerkvig R, Deutsch A, Voss-Bohme A, Niclou SP (2019) Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun 10(1):1787. https://doi.org/10.1038/s41467-019-09853-z
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11(1):69–82. https://doi.org/10.1016/j.ccr.2006.11.020
Shweiki D, Itin A, Soffer D, Keshet E (1992) Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359(6398):843–845. https://doi.org/10.1038/359843a0
Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, Simons M, Tam W, Felsher DW, Shido K, Rafii A, Scandura JM, Rafii S (2014) Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 25(3):350–365. https://doi.org/10.1016/j.ccr.2014.02.005
Pasquier J, Ghiabi P, Chouchane L, Razzouk K, Rafii S, Rafii A (2020) Angiocrine endothelium: from physiology to cancer. J Transl Med 18(1):52. https://doi.org/10.1186/s12967-020-02244-9
Kumar S, Sharife H, Kreisel T, Mogilevsky M, Bar-Lev L, Grunewald M, Aizenshtein E, Karni R, Paldor I, Shlomi T, Keshet E (2019) Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab 30(1):201-211e206
Rafii S, Butler JM, Ding BS (2016) Angiocrine functions of organ-specific endothelial cells. Nature 529(7586):316–325. https://doi.org/10.1038/nature17040
Weber JM, Calvi LM (2010) Notch signaling and the bone marrow hematopoietic stem cell niche. Bone 46(2):281–285. https://doi.org/10.1016/j.bone.2009.08.007
Brooks LJ, Parrinello S (2017) Vascular regulation of glioma stem-like cells: a balancing act. Curr Opin Neurobiol 47:8–15. https://doi.org/10.1016/j.conb.2017.06.008
Matsumoto K, Arao T, Tanaka K, Kaneda H, Kudo K, Fujita Y, Tamura D, Aomatsu K, Tamura T, Yamada Y, Saijo N, Nishio K (2009) mTOR signal and hypoxia-inducible factor-1 alpha regulate CD133 expression in cancer cells. Cancer Res 69(18):7160–7164. https://doi.org/10.1158/0008-5472.CAN-09-1289
Yan GN, Yang L, Lv YF, Shi Y, Shen LL, Yao XH, Guo QN, Zhang P, Cui YH, Zhang X, Bian XW, Guo DY (2014) Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol 234(1):11–22. https://doi.org/10.1002/path.4349
Fessler E, Borovski T, Medema JP (2015) Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol Cancer 14:157. https://doi.org/10.1186/s12943-015-0420-3
Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, Cuartas I, Raventos C, Martinez-Ricarte F, Poca MA, Garcia-Dorado D, Lahn MM, Yingling JM, Rodon J, Sahuquillo J, Baselga J, Seoane J (2010) TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18(6):655–668. https://doi.org/10.1016/j.ccr.2010.10.023
Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He X, Hervey-Jumper SL, Heth JA, Muraszko KM, DiMeco F, Vescovi AL, Fan X (2011) Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res 71(18):6061–6072. https://doi.org/10.1158/0008-5472.CAN-10-4269
Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, Correia AS, Soulet D, Major T, Menon J, Tabar V (2010) Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 28(6):1019–1029. https://doi.org/10.1002/stem.429
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109. https://doi.org/10.1007/s00401-007-0243-4
Kumar S, Sharife H, Kreisel T, Bar-Lev L, Grunewald M, Keshet E (2020) Isolation of tumor cells based on their distance from blood vessels. Bio-Protocol. https://doi.org/10.21769/BioProtoc.3628
Hu Y, Smyth GK (2009) ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347(1–2):70–78. https://doi.org/10.1016/j.jim.2009.06.008
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31(1):46–53. https://doi.org/10.1038/nbt.2450
Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, Dor Y, Regev A, Yanai I (2016) CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol 17:77. https://doi.org/10.1186/s13059-016-0938-8
Taverna F, Goveia J, Karakach TK, Khan S, Rohlenova K, Treps L, Subramanian A, Schoonjans L, Dewerchin M, Eelen G, Carmeliet P (2020) BIOMEX: an interactive workflow for (single cell) omics data interpretation and visualization. Nucleic Acids Res 48(W1):W385–W394. https://doi.org/10.1093/nar/gkaa332
Kim WT, Ryu CJ (2017) Cancer stem cell surface markers on normal stem cells. BMB Rep 50(6):285–298. https://doi.org/10.5483/bmbrep.2017.50.6.039
Richichi C, Brescia P, Alberizzi V, Fornasari L, Pelicci G (2013) Marker-independent method for isolating slow-dividing cancer stem cells in human glioblastoma. Neoplasia 15(7):840–847. https://doi.org/10.1593/neo.13662
Civenni G, Walter A, Kobert N, Mihic-Probst D, Zipser M, Belloni B, Seifert B, Moch H, Dummer R, van den Broek M, Sommer L (2011) Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res 71(8):3098–3109. https://doi.org/10.1158/0008-5472.CAN-10-3997
Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ, Longaker MT, Weissman IL (2010) Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 466(7302):133–137. https://doi.org/10.1038/nature09161
Imai T, Tamai K, Oizumi S, Oyama K, Yamaguchi K, Sato I, Satoh K, Matsuura K, Saijo S, Sugamura K, Tanaka N (2013) CD271 defines a stem cell-like population in hypopharyngeal cancer. PLoS ONE 8(4):e62002. https://doi.org/10.1371/journal.pone.0062002
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9(6):400–414. https://doi.org/10.1038/nrc2657
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13(12):1501–1512. https://doi.org/10.1101/gad.13.12.1501
Engeland K (2018) Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ 25(1):114–132. https://doi.org/10.1038/cdd.2017.172
Alshehri MM, Robbins SM, Senger DL (2017) The role of neurotrophin signaling in gliomagenesis: a focus on the p75 neurotrophin receptor (p75(NTR)/CD271). Vitam Horm 104:367–404
Johnston AL, Lun X, Rahn JJ, Liacini A, Wang L, Hamilton MG, Parney IF, Hempstead BL, Robbins SM, Forsyth PA, Senger DL (2007) The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol 5(8):e212. https://doi.org/10.1371/journal.pbio.0050212
Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K, Tofilon PJ (2014) The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol 16(1):29–37. https://doi.org/10.1093/neuonc/not139
Sunayama J, Matsuda K, Sato A, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A, Kitanaka C (2010) Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 28(11):1930–1939. https://doi.org/10.1002/stem.521
Sunayama J, Sato A, Matsuda K, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A, Kitanaka C (2010) Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro Oncol 12(12):1205–1219. https://doi.org/10.1093/neuonc/noq103
O’Brien C, Cavet G, Pandita A, Hu X, Haydu L, Mohan S, Toy K, Rivers CS, Modrusan Z, Amler LC, Lackner MR (2008) Functional genomics identifies ABCC3 as a mediator of taxane resistance in HER2-amplified breast cancer. Cancer Res 68(13):5380–5389. https://doi.org/10.1158/0008-5472.CAN-08-0234
Calatozzolo C, Gelati M, Ciusani E, Sciacca FL, Pollo B, Cajola L, Marras C, Silvani A, Vitellaro-Zuccarello L, Croci D, Boiardi A, Salmaggi A (2005) Expression of drug resistance proteins Pgp, MRP1, MRP3, MRP5 and GST-pi in human glioma. J Neurooncol 74(2):113–121. https://doi.org/10.1007/s11060-004-6152-7
Yin S, Xu L, Bonfil RD, Banerjee S, Sarkar FH, Sethi S, Reddy KB (2013) Tumor-initiating cells and FZD8 play a major role in drug resistance in triple-negative breast cancer. Mol Cancer Ther 12(4):491–498. https://doi.org/10.1158/1535-7163.MCT-12-1090
Ferrari-Toninelli G, Bonini SA, Uberti D, Buizza L, Bettinsoli P, Poliani PL, Facchetti F, Memo M (2010) Targeting notch pathway induces growth inhibition and differentiation of neuroblastoma cells. Neuro Oncol 12(12):1231–1243. https://doi.org/10.1093/neuonc/noq101
Ottone C, Krusche B, Whitby A, Clements M, Quadrato G, Pitulescu ME, Adams RH, Parrinello S (2014) Direct cell-cell contact with the vascular niche maintains quiescent neural stem cells. Nat Cell Biol 16(11):1045–1056. https://doi.org/10.1038/ncb3045
Sestan N, Artavanis-Tsakonas S, Rakic P (1999) Contact-dependent inhibition of cortical neurite growth mediated by notch signaling. Science 286(5440):741–746
Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, Sundaresan T, Pastorino S, Park JK, Mikolaenko I, Maric D, Eberhart CG, Fine HA (2005) Expression of notch-1 and its ligands, delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res 65(6):2353–2363
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715. https://doi.org/10.1016/j.cell.2008.03.027
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9(4):265–273. https://doi.org/10.1038/nrc2620
Teodorczyk M, Schmidt MHH (2014) Notching on cancer’s door: notch signaling in brain tumors. Front Oncol 4:341. https://doi.org/10.3389/fonc.2014.00341
Chen J, Kesari S, Rooney C, Strack PR, Chen J, Shen H, Wu L, Griffin JD (2010) Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 1(8):822–835. https://doi.org/10.1177/1947601910383564
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL, Eberhart CG (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28(1):5–16. https://doi.org/10.1002/stem.254
Xie Q, Wu Q, Kim L, Miller TE, Liau BB, Mack SC, Yang K, Factor DC, Fang X, Huang Z, Zhou W, Alazem K, Wang X, Bernstein BE, Bao S, Rich JN (2016) RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J Clin Invest 126(7):2757–2772. https://doi.org/10.1172/JCI86114
Schreck KC, Taylor P, Marchionni L, Gopalakrishnan V, Bar EE, Gaiano N, Eberhart CG (2010) The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin Cancer Res 16(24):6060–6070. https://doi.org/10.1158/1078-0432.CCR-10-1624
Bazzoni R, Bentivegna A (2019) Role of notch signaling pathway in glioblastoma pathogenesis. Cancers (Basel). https://doi.org/10.3390/cancers11030292
Bayin NS, Frenster JD, Sen R, Si S, Modrek AS, Galifianakis N, Dolgalev I, Ortenzi V, Illa-Bochaca I, Khahera A, Serrano J, Chiriboga L, Zagzag D, Golfinos JG, Doyle W, Tsirigos A, Heguy A, Chesler M, Barcellos-Hoff MH, Snuderl M, Placantonakis DG (2017) Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget 8(39):64932–64953. https://doi.org/10.18632/oncotarget.18117
Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, Deli MA, Forster C, Galla HJ, Romero IA, Shusta EV, Stebbins MJ, Vandenhaute E, Weksler B, Brodin B (2016) In vitro models of the blood-brain barrier: an overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab 36(5):862–890. https://doi.org/10.1177/0271678X16630991
Man S, Ubogu EE, Williams KA, Tucky B, Callahan MK, Ransohoff RM (2008) Human brain microvascular endothelial cells and umbilical vein endothelial cells differentially facilitate leukocyte recruitment and utilize chemokines for T cell migration. Clin Dev Immunol 2008:384982. https://doi.org/10.1155/2008/384982
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, Varambally S (2017) UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19(8):649–658. https://doi.org/10.1016/j.neo.2017.05.002
Acknowledgements
We thank Dr.Sharona Elgavish and the Info-Core Bioinformatics unit of the Hebrew University faculty of Medicine for bioinformatics analysis, all the members of the Keshet lab for their inputs and Dr. Aparna Anand for proof reading the manuscript. This work was supported by the Cooperation Program in Cancer Research of the Deutsches Krebsforschungszentrum (DKFZ), the Israeli Ministry of Science and Technology (MOST) to EK (CA-178) and New Faculty Grant from Indian Institute of Technology Delhi to SK (MI00148).
Disclosure
The authors declare that no competing interests exist.
Author information
Authors and Affiliations
Contributions
SK conceived, designed, and performed experiments. LB performed in vitro co-culture experiments and data analysis. MM, HS, MG, and TL assisted in animal studies. FT, JG, and PC analyzed transcriptomics/metabolomics data of co-culture experiments. EK conceived and supervised the study. SK and EK wrote the manuscript.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 (MP4 17918 kb)
Rights and permissions
About this article

Cite this article
Kumar, S., Bar-Lev, L., Sharife, H. et al. Identification of vascular cues contributing to cancer cell stemness and function. Angiogenesis 25, 355–371 (2022). https://doi.org/10.1007/s10456-022-09830-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-022-09830-z