
Abstract
Using density functional theory (DFT), we presented a theoretical investigation of CO and NO gas molecules adsorption on the Al-doped, P-doped and Si-doped MoS2 monolayers. Our main focus is on the interactions between the dopants (Al, P and Si) and gas molecules. The properties of the adsorption system were analyzed in view of the density of states, electron density distribution, charge density differences and electronic band structures. Various orientations of CO and NO molecules were considered on the MoS2 monolayer to search for the most stable configurations. The results suggest that the adsorption of gas molecules on the doped MoS2 monolayers is more favorable in energy than that on the pristine monolayers. This means that the interaction between doped MoS2 and gas molecules is stronger than that between pristine MoS2 and gas molecules. Our calculations show shorter adsorption distance and higher adsorption energy for Al-doped and Si-doped monolayers than the P-doped and pristine ones. Charge density difference calculations show the charge accumulation between the interacting atoms, suggesting the formation of covalent bonds, as evidenced by the projected density of states of the interacting atoms. Our results confirm that Al-doped and Si-doped MoS2 can be used as efficient and promising sensor materials for CO and NO detection in the environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
A promising gas sensor, for the detection of harmful gas molecules, plays an important role for public health, environmental protection, agricultural and biomedical processes. In the past decades, the emerging two-dimensional layered materials, such as graphene, have received great attentions because of their efficient gas sensing characteristics (Yuan and Shi 2013; Kemp et al. 2013; Huang et al. 2008; Leenaerts et al. 2008). It has been demonstrated that the substitutional doping of graphene with some metal and nonmetal elements is a successful strategy to amend and enhance the interaction between the substrate and adsorbates, which greatly improves the electronic properties of carbon based nanomaterials (Zhou et al. 2009). Thus, the chemical doping promotes and increases the sensing properties of graphene based gas sensors (Kang et al. 2013; Niu et al. 2013; Dai et al. 2009; Ao et al. 2010). Despite to the graphene, two-dimensional layered dichalcogenides such as MoS2 are receiving significant interests in the field of gas sensors, and proposed as the effective materials for sensing elements due to their excellent electrical, optical, and catalytic properties (Wang et al. 2012; Wilcoxon et al. 1999; Sienicki and Hryniewicz 1996; Fernandez-Garcia et al. 2004). Among the various 2D layered materials, MoS2 monolayers, as one of the most important transition-metal dichalcogenides have attracted widespread interest in numerous research fields, which possess excellent properties such as good electrical transport, excellent chemical and thermal stability, and high surface-to-volume ratio (Chen et al. 2019; Zhang et al. 2017; Cho et al. 2015; Fan et al. 2017). In this regard, the stability of the MoS2 monolayers plays a key role. Since the Al, Si, P, and S atoms have the same electronic configuration, and the covalent radii of these atoms are close to each other, the stable covalent structures of Al, P and Si dopants into the S vacancy of MoS2 monolayer can be easily formed. Luo et al. calculated the formation energies for the introduction of dopant atoms and suggested that Al, P and Si-doped MoS2 monolayers are thermodynamically stable with reasonable formation energies (Luo et al. 2016). The formation energies of Al, P and Si atoms into the MoS2 sheet are 2.66, 1.07 and 0.61 eV. Therefore, they concluded that these doped structure are also thermally stable. Dai et al. studied the adsorption of NO and NO2 gas molecules on B, N, Al and S-doped graphene, and suggested that the chemical doping improves the sensing performance of the materials (Niu et al. 2013). Niu et al. fabricated the N-Si codoped graphene nanosheets in the laboratory, and examined the NO2 sensing behaviors of the nanosheets (Dai et al. 2009). The adsorption of different gas molecules on the surface of 2D materials and metal oxide nanoparticles have been also investigated in our previous works (Abbasi and Sardroodi 2016a, b, 2017a, b, 2018a, b, c, 2019a, b, Abbasi 2019). As inspired by the advent of graphene-FET sensors, the 2D dichalcogenides have aroused enormous interests as emerging sensing materials. Similar to graphene, the high surface to volume ratio of dichalcogenide monolayers makes them efficient candidates for gas sensing applications (Yue et al. 2013; Li et al. 2012; Zhao et al. 2014; Late et al. 2013; Donarelli et al. 2015; He et al. 2012; Cho et al. 2015). The adsorption of nitrogen oxides and ammonia molecules on graphene and graphene oxide surfaces have been systematically studied by Tang and Cao (2011, 2012). Of the 2D dichalcogenides, MoS2 monolayers show high sensitivity toward some important gas molecules, such as NOx and NH3 in the environment (Donarelli et al. 2015; Lee et al. 2013; Late et al. 2013; Yao et al. 2013). It is very important to the chemical sensor community that the semiconducting material is required for the sensing characteristics of gas molecules. However, in the case of graphene monolayer, the band gap is zero, which limits its sensing performance. Comparing with graphene, MoS2 exhibits better semiconducting nature with the band gap in the range of 1.3–1.8 eV (Huang et al. 2013). Furthermore, owing to the semiconductor characteristics of MoS2 nanosheets, the transport properties and sensing capabilities can be tuned by applying gate voltages (Late et al. 2013). Therefore, it is of eminent significance to examine the adsorption of gas molecules on the perfect and modified MoS2 monolayers. It is well-known that NO and CO are harmful air pollutants with detrimental effects on the living organisms. Hence, detecting and monitoring of these pollutants in the atmosphere are indispensable to reduce the concentration of such contaminants to an environmentally acceptable value. In this work, we carried out density functional theory calculations to explore the adsorption of gas molecules such as CO and NO on the pristine, Al-doped, P-doped and Si-doped MoS2 monolayers. We conducted a systematic theoretical investigation of the structure, adsorption energies, electronic band structures of Al-, Si-, and P-doped MoS2 monolayers, and analysis of the charge density difference and density of states of the studied systems. The interactions of doped MoS2 monolayers with CO and NO gas molecules are analyzed in detail so as to provide theoretical insights into the electronic properties of doped MoS2 based gas sensors.
2 Methods and calculation models
The total energy and electronic structure calculations based on density functional theory (DFT) (Hohenberg and Kohn 1964; Kohn and Sham 1965) were performed using the SIESTA code (Soler et al. 2002). The double-zeta polarized (DZP) basis sets were used to expand the electronic density with norm-conserving Trouiller–Martins pseudopotential (Troullier and Martins 1991). Graphical Display Interface for Structures (GDIS) program was employed to manipulate and model the periodic systems such as stanene supercells (Koklj 2003). The convergence tolerance for total energy was set to 10−4 eV, and all the systems were geometrically optimized using the conjugate gradient (CG) method. Moreover, the Brillouin zone (BZ) integration was performed by setting a 10 × 10 × 1 Monkhorst–Pack k-point grid for a 4 × 4 × 1 supercell (Monkhorst and Pack 1976). The generalized gradient approximation (GGA) potential (Blochl 1994; Kresse and Joubert 1999), parameterized by the Perdew–Burke–Ernzerhof (PBE) was used for the exchange–correlation functional (Perdew et al. 1997) to perform the total energy calculations and geometry relaxations. VESTA (visualization for electronic and structural analysis) program was employed for the display of the charge density difference and total electron density plots (Momma and Izumi 2011). The unit cell of MoS2 monolayer was extended to a 4 × 4 × 1 supercell containing 48 atoms (16 Mo and 32 S atoms). To characterize the stability of the adsorption configurations, CO and NO molecules were placed at different positions and sites towards the MoS2 monolayer. The adsorption energy is calculated as follows, Eq. (1):
where \(E_{{(MoS_{2} \, + \, gas \, molecule)}}\) is the total energy of the MoS2 monolayer with adsorbed gas molecule, and \(E_{{MoS_{2} }}\) is the total energy of the bare MoS2 monolayer, \(E_{gas \, molecule}\) is the total energy of isolated gas molecule. Based on the above equation, the more negative the adsorption energy, the higher the stability of the configuration.
3 Results and discussion
3.1 Structural and electronic properties of doped MoS2 monolayers
We initially optimized the structure of the pristine MoS2 monolayer, and introduced one of the dopants (Al, P and Si) into the sulfur vacancy of the MoS2. The optimized structures of the pristine, Al-doped, P-doped and Si-doped MoS2 monolayers were displayed in Fig. 1a–d. The calculated Mo-S bond length and Mo–Mo distance for pristine MoS2 are 2.45 Å and 3.19 Å, respectively, which are in reasonable agreement with the theoretical (Le et al. 2014; Ataca and Ciraci 2011) and experimental data (Joensen et al. 1987). Moreover, based on the optimized structures, the Mo-Al, Mo-P and Mo-Si bond lengths were calculated to be 2.55 Å, 2.42 Å and 2.38 Å, respectively, which are very close to the reported values (Luo et al. 2016). We conducted this work following the computational study of Ma et al. (2016). In order to further investigate the electronic properties of the doped MoS2 systems, we have calculated their electronic band structures. The band structure plots were depicted in Fig. 2. As can be seen, pristine MoS2 exhibits a semiconductor characteristics with a calculated direct band gap of about 1.71 eV (Fig. 2a), which is consistent with the previous reports (Cheng et al. 2013; Mak et al. 2010). Besides, the Al-doped, P-doped and Si-doped systems show semiconducting behavior. Thus, the doped MoS2 monolayers can be used as efficient gas sensors for detecting CO and NO molecules. For Al-doped system (Fig. 2b), a band state located at the energy value of about − 3.8 eV could be ascribed to the Al atom, while in the case of Si-doped system (Fig. 2d), this state appears at − 4.1 eV, which mainly comes from the Si atom. The P-doped MoS2 exhibits an interesting behavior with a Fermi level touching the conduction band minimum (CBM). Therefore, the P-doped system could also be utilized for sensing gas molecules because of its semiconductor behavior.

Optimized structures of the pristine (a), Al-doped (b), P-doped (c) and Si-doped (d) MoS2 monolayers

Band structure plots of the pristine (a), Al-doped (b), P-doped (c) and Si-doped (d) MoS2 monolayers
3.2 Adsorption of CO molecule on the doped MoS2 monolayers
The results of CO adsorption on the pristine, Al-doped, P-doped and Si-doped MoS2 monolayers were presented in this section. For the adsorption of CO molecule on the doped MoS2 monolayer, various adsorption orientations were tested to obtain the most stable configuration. The optimized structures of the CO adsorbed pristine and doped MoS2 monolayers in different views were displayed in Figs. 3 and 4. Both carbon and oxygen atoms of the CO molecule were considered to be adsorption sites (Ma et al. 2016). Our results suggest that the perpendicular adsorption of CO on the MoS2 is more energetically favorable than the parallel adsorption, and gives rise to the most stable structures. Thus, we have only presented the perpendicular adsorption configurations. A brief description of the CO adsorbed MoS2 configurations is as follows: (A) CO molecule is positioned towards the pristine MoS2 with carbon atom at the top of the sulfur site of MoS2; (B) The same configuration with oxygen atom positioned at the top of the sulfur site; (C) CO molecule is located above the Al-doped MoS2 with carbon atom at the top of the Al site; (D) CO molecule adsorbed on the surface with oxygen atom oriented above the Al site of doped MoS2. Configurations A and B represent a weak physisorption of CO on the MoS2 surface either from carbon or from oxygen site of CO molecule. There is no covalent bond between CO molecule and pristine MoS2 monolayer, as confirmed by the total electron density profiles (Fig. 5). In contrast, configurations C and D show the strong chemisorption of CO molecule on the Al-doped MoS2 monolayer with considerable adsorption energies. Both carbon and oxygen atoms of the CO molecule form strong chemical bond with the impurity Al atom of doped MoS2 after the adsorption process (Ma et al. 2016). Thus, the adsorption distances for configurations C and D are lower than those of configurations A and B, which lead to the higher adsorption energies of configurations C and D. The calculated bond length of the free CO is 1.14 Å. From Table 1, the C-O bond length of the adsorbed CO molecule is slightly increased by 0.01–0.03 Å. This suggests that the C–O bond length is stretched after the adsorption process. These smaller distances between the adsorbed CO molecule and the doped Al/Si atoms support the strong interaction between the adsorbed molecule and MoS2 monolayers. We found that the variation trend of the CO-dopant distance is well correlated with that of the adsorption energies. The adsorption energy analysis was also conducted to evaluate the stability of the CO adsorbed MoS2 monolayers. The results were summarized in Table 1. As can be seen from this Table, the adsorption energies of configurations C and D (Al-doped MoS2) are higher than those of configurations A and B (pristine MoS2), which are mostly attributed to the chemical nature of adsorption in configurations C and D. However, the lower adsorption energies of configurations A and B confirm the weak interaction between CO and pristine MoS2 monolayer. Therefore, CO adsorption on the Al-doped MoS2 is more favorable in energy than that on the pristine one. To achieve further insights into the interaction between the adsorbed CO and the doped MoS2 monolayers, we have calculated the projected density of states (PDOS) profiles of the Al atom and C/O atoms of the CO molecule (Fig. 5). As can be seen from this Figure, there are large overlaps between the PDOS profiles of the Al atom and C/O atoms of CO molecule, indicating the formation of covalent bonds between these atoms.

Optimized configurations of CO molecule adsorbed on the pristine and Al-doped MoS2 monolayers. The green, yellow, red and gray balls represent Mo, S, O and C atoms, respectively. Subfigures a–d represent configurations A–D (Color figure online)

Optimized configurations of CO molecule adsorbed on the P-doped and Si-doped MoS2 monolayers. The green, yellow, red and gray balls represent Mo, S, O and C atoms, respectively. Subfigures e–h represent configurations E–H (Color figure online)

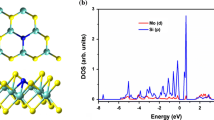
Projected density of states of the Al, Si, C and O atoms for CO adsorption on the Al-doped and Si-doped MoS2 monolayers along with the total electron density distribution plots. The isosurface value of electron density is taken as 0.002 e/Å3
The electronic properties of the adsorption systems were also described in terms of energy band structure. Figure 6 presents the band structure plots of the pristine and doped MoS2 monolayers with adsorbed CO molecules. Figure 6a and b represents the band structure plots for configurations A and B. As can be seen from this Figure, the electronic structure of the pristine MoS2 monolayer remains almost unaltered after the adsorption of CO molecule on its surface (Ma et al. 2016). The biggest difference between the band structures of the pristine MoS2 and CO adsorbed MoS2 is the slight shift in the position of the Fermi level. The band structure plots of the Al-doped MoS2 after the adsorption process (configurations C and D) were also displayed in Fig. 6c and d. This Figure shows that the electronic structure of Al-doped MoS2 was changed after the adsorption of CO molecule, which can be attributed to the formation of chemical bonds between the Al atom and C/O atom of CO molecule. The energy band structure plots of the P-doped and Si-doped MoS2 after CO adsorption were also depicted in Fig. 6e and h. In configurations E and F, CO molecules weakly interact with P-doped MoS2 monolayer, and this weak interaction gives rise to a slight change in the band structure of the complex system. The band structure plots of the configurations G and H (CO adsorbed Si-doped MoS2 system) exhibit significant changes compared to the band structure plots of non-adsorbed systems. For Al-doped and Si-doped MoS2 monolayers, CO adsorption induces impurity states inside the band gap of MoS2, which is mostly attributed to the formation of covalent bonds between interacting atoms (Ma et al. 2016).

Band structure plots of the pristine, Al-doped, P-doped and Si-doped MoS2 monolayers with adsorbed CO molecules. The Fermi level is indicated by a cyan solid line
The changes in the electronic structure induced by the CO adsorption were also analyzed in view of the charge density difference (CDD) calculations for CO adsorbed Al-embedded and Si-embedded MoS2 monolayers. Figure 7 represents the CDD plots for the considered systems. The charge density difference is a measure of the difference between the electron densities of the non-interacting systems (CO/NO molecule and MoS2 monolayer) and the complex system consisting of MoS2 with adsorbed gas molecules. The atomic coordinates of isolated gas molecule and MoS2 monolayer would be the same as those in the complex MoS2 + gas system. The charge density difference was evaluated based on the following equation:
where, ρ(MoS2 + gas molecule) and ρ(MoS2), are the total electron densities of the gas adsorbed and pristine MoS2 monolayers, respectively and ρ(gas molecule) represents the electron density of the free gas molecule. In the figure, the yellow and cyan regions for the CDD represent the areas of electron accumulation and depletion, respectively. As can be seen from Fig. 7, there is some electron accumulation on the adsorbed CO molecule. The electron accumulation between the interacting C/O and Al/Si atoms is significant, indicating the covalent interaction between the adsorbed CO molecule and the Al/Si-doped MoS2 monolayer. This increase of the electronic density confirms the formation of chemical Al-C/Al-O and Si–C/Si–O bonds and consequently chemisorption of CO molecule on the Al-embedded and Si-embedded monolayers. The strong interaction between CO and the Al/Si doped monolayer is also evidenced by the PDOS spectra shown in Fig. 5, where the substantial overlapping is clear between the PDOS states of the C/O atoms of the adsorbed CO and the dopants.

Charge density difference plots for CO adsorbed Al-doped and Si-doped MoS2 monolayers. The isosurface value is taken as 0.002 e/Å3
3.3 Adsorption of NO molecule on the MoS2 monolayers
For NO adsorption on the pristine and doped MoS2 monolayers, we have considered various adsorption configurations. Optimized geometry configurations of the pristine and doped MoS2 with adsorbed NO molecules were shown in Figs. 8 and 9. For each pair, the first configuration represents the adsorption of CO on the monolayer by its N site, while the second one shows the adsorption through its O site. As can be seen from Fig. 8, configurations A and B represent the interaction of NO molecule with MoS2 through its N and O atoms, respectively. Similarly, there is a weak mutual interaction between pristine MoS2 and NO molecule. In contrast, Al-doped and Si-doped system strongly interacts with NO molecule (configurations C and D). The adsorption of NO molecule on the P-doped and Si-doped MoS2 monolayers were also depicted ac configurations E-F and G-H for P-doped and Si-doped systems, respectively. It can be seen from these Figures that the NO molecule chemisorbs on the Al-doped and Si-doped MoS2 monolayers, as evidenced by the total electron density distribution plots (Fig. 10). Similar to the pristine MoS2, P-doped system shows a weak physisorption above the monolayer surface. The distances between NO and the Al/Si-doped MoS2 are very short compared to those between NO and pristine one, suggesting the strong interaction between the NO molecule and the Al/Si-doped MoS2 monolayers (Ma et al. 2016). Important to note is that the change trend of the distances between the adsorbed NO molecule and MoS2 monolayer is consistent with that of the adsorption energies. The calculated bond length of the isolated NO molecule is 1.16 Å. As can be seen from Table 2, the bond length of the NO molecule is slightly stretched by the adsorption process. The adsorption energy values were also listed in Table 2. The results suggest that the adsorption of NO molecule on the doped MoS2 monolayers is more energetically favorable than that on the pristine ones. The higher adsorption energies of the Al-doped and Si-doped systems reveal a strong adsorption of NO molecule on the Al and Si sites. Thus, Al/P/Si embedding improves the interaction between gas molecule and MoS2 monolayer. In order to further discuss the chemisorption of NO molecule on the Al-embedded and Si-embedded systems, we have calculated their projected density of states (Fig. 10). As can be seen from this figure, the significant overlaps between the PDOS spectra of the N/O and Al/Si atoms indicate the formation of chemical bonds between them. Similar with CO adsorption, we considered the energy band structure plots for NO adsorbed on the pristine and doped MoS2 monolayers, as represented in Fig. 11. It can be seen from this figure that the electronic structures of the doped MoS2 systems show significant changes after NO adsorption including the creation of some states inside the band gap of MoS2. Therefore, NO adsorption alters the electronic band structure of doped MoS2 monolayers by changing the position of Fermi level and introducing impurity states in the band structure of MoS2 (Ma et al. 2016).

Optimized configurations of NO molecule adsorbed on pristine and the Al-doped MoS2 monolayers. The green, yellow, red and gray balls represent Mo, S, O and C atoms, respectively (Color figure online)

Optimized configurations of NO molecule adsorbed on the P-doped and Si-doped MoS2 monolayers. The green, yellow, red and gray balls represent Mo, S, O and C atoms, respectively (Color figure online)

Projected density of states of the Al, Si, C and O atoms for NO adsorption on the Al-doped and Si-doped MoS2 monolayers along with the total electron density distribution plots. The isosurface value of electron density is taken as 0.002 e/Å3

Band structure plots of the pristine, Al-doped, P-doped and Si-doped MoS2 monolayers with adsorbed NO molecules. The Fermi level is indicated by a cyan solid line
We then focused our attention on the charge density difference (CDD) plots for NO adsorption on the MoS2 monolayers (Fig. 12). This figure shows that for Al-embedded and Si-embedded MoS2 systems, there is some electron accumulation on the adsorbed NO molecule. Besides, the higher accumulation of electron density between the N/O atoms of the NO and the doped Al/Si atoms indicates the covalent characteristics for the interaction between NO and the Al/Si-doped MoS2 monolayers and consequently the chemical nature of the adsorption.

Charge density difference plots for NO adsorbed Al-doped and Si-doped MoS2 monolayers. The isosurface value is taken as 0.002 e/Å3
4 Conclusions
To examine the possible application of MoS2 monolayers in gas sensing, the adsorption of CO and NO molecules on the MoS2 monolayers embedded with Al, P and Si atoms were investigated using the density functional theory calculations. Various adsorption positions of gas molecules on the pristine and doped monolayers were thoroughly examined. Based on our calculated results, CO and NO molecules show a weak physisorption on the pristine MoS2 monolayer, whereas the doped systems (especially Al-doped and Si-doped) exhibit a strong chemisorption on the surface. Adsorption energy results indicate that the adsorption of gas molecules on the doped systems is more favorable in energy than that on the pristine ones. For gas adsorption on the Al-doped and Si-doped MoS2, the large overlaps between the PDOS spectra of the Al/Si and C/O/N atoms represent the formation of chemical bonds between them. The energy band structure plots also show that the electronic structure of the Al-doped and Si-doped monolayers were considerably altered upon gas adsorption, while in the case of pristine monolayer, the electronic structure remains almost unaltered. The charge density difference plots also indicate the accumulation of electron density between the interacting atoms and consequently formation of covalent bonds between them. Finally, the present work shows that embedding impurity atoms may be a useful strategy to tune the adsorption behaviors of MoS2 monolayers.
References
Abbasi, A.: Adsorption of phenol, hydrazine and thiophene on stanene monolayers: a computational investigation. Synth. Metal 247, 26–36 (2019)
Abbasi, A., Sardroodi, J.J.: N-doped TiO2 anatase nanoparticles as a highly sensitive gas sensor for NO2 detection: insights from DFT computations. Environ. Sci. Nano 3, 1153–1164 (2016a)
Abbasi, A., Sardroodi, J.J.: Theoretical study of the adsorption of NOx on TiO2/MoS2 nanocomposites: a comparison between undoped and N-doped nanocomposites. J. Nanostruct. Chem. 6, 309–327 (2016b)
Abbasi, A., Sardroodi, J.J.: An innovative gas sensor system designed from a sensitive nanostructured ZnO for the selective detection of SOx molecules: a density functional theory study. N. J. Chem. 41, 12569–12580 (2017a)
Abbasi, A., Sardroodi, J.J.: Prediction of a highly sensitive molecule sensor for SOx detection based on TiO2/MoS2 nanocomposites: a DFT study. J. Sulfur Chem. 38(1), 52–68 (2017b)
Abbasi, A., Sardroodi, J.J.: Investigation of the adsorption of ozone molecules on TiO2/WSe2 nanocomposites by DFT computations: applications to gas sensor devices. Appl. Surf. Sci. 436, 27–41 (2018a)
Abbasi, A., Sardroodi, J.J.: Structural and electronic properties of group-IV tin nanotubes and their effects on the adsorption of SO2 molecules: insights from DFT computations. J. Appl. Phys. 124, 165302 (2018b)
Abbasi, A., Sardroodi, J.J.: Exploration of sensing of nitrogen dioxide and ozone molecules using novel TiO2/Stanene heterostructures employing DFT calculations. Appl. Surf. Sci. 442, 368–381 (2018c)
Abbasi, A., Sardroodi, J.J.: The adsorption of sulfur trioxide and ozone molecules on stanene nanosheets investigated by DFT: applications to gas sensor devices. Physica E 108, 382–390 (2019a)
Abbasi, A., Sardroodi, J.J.: Adsorption of O3, SO2 and SO3 gas molecules on MoS2 monolayers: a computational investigation. Appl. Surf. Sci. 469, 781–791 (2019b)
Ao, Z.M., Li, S., Jiang, Q.: Correlation of the applied electrical field and CO adsorption/desorption behavior on Al-doped graphene. Solid State Commun. 150, 680 (2010)
Ataca, C., Ciraci, S.: Functionalization of single-layer MoS2 honeycomb structures. J. Phys. Chem. C 115, 13303–13311 (2011)
Blochl, P.E.: Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994)
Chen, D.C., Tang, J., Zhang, X.X., Li, Y., Liu, H.J.: Detecting decompositions of sulfur hexafluoride using MoS2 Monolayer as gas sensor. IEEE Sens. J. 19, 39–46 (2019)
Cheng, Y.C., Zhu, Z.Y., Mi, W.B., Guo, Z.B., Schwingenschlogl, U.: Prediction of two-dimensional diluted magnetic semiconductors: doped monolayer MoS2 systems. Phys. Rev. BV 87, 100401 (2013)
Cho, B., Yoon, J., Lim, S.K., Kim, A.R., Kim, D.H., Park, S.G., Kwon, J.D., Lee, Y.J., Lee, K.H., Lee, B.H.: Chemical sensing of 2D graphene/MoS2 heterostructure device. ACS Appl. Mater. Int. 7, 16775–16780 (2015a)
Cho, B., Hahm, M.G., Choi, M., Yoon, J., Kim, A.R., Lee, Y.-J., Park, S.-G., Kwon, J.-D., Kim, C.S., Song, M., Jeong, Y., Nam, K.-S., Lee, S., Yoo, T.J., Kang, C.G., Lee, B.H., Ko, H.C., Ajayan, P.M., Kim, D.-H.: Charge-transfer-based Gas sensing using atomic-layer MoS2. Sci. Rep. 5, 8052 (2015b)
Dai, J., Yuan, J., Giannozzi, P.: Gas adsorption on graphene doped with B, N, Al, and S: A theoretical study. Appl. Phys. Lett. 95, 232105 (2009)
Donarelli, M., Prezioso, S., Perrozzi, F., Bisti, F., Nardone, M., Giancaterini, L., Cantalini, C., Ottaviano, L.: Response to NO2 and other gases of resistive chemically exfoliated MoS2-based gas sensors. Sens. Actuat. B 207, 602–613 (2015)
Fan, C., Liu, G.Z., Zhang, Y.H., Wang, M.J.: Synthesis and gas-responsive characteristics to methanol and isopropanol of bean-sprout-like MoS2. Mater. Lett. 209, 8–10 (2017)
Fernandez-Garcia, M., Martinez-Arias, A., Hanson, J.C., Rodriguez, J.A.: Nanostructured oxides in chemistry: characterization and properties. J. Chem. Rev. 104, 4063–4104 (2004)
He, Q., Zeng, Z., Yin, Z., Li, H., Wu, S., Huang, X., Zhang, H.: Fabrication of flexible MoS2 thin-film transistor arrays for practical gas-sensing applications. Small 8, 2994 (2012)
Hohenberg, P., Kohn, W.: Inhomogeneous Electron gas. Phys. Rev. 136, B864–B871 (1964)
Huang, B., Li, Z., Liu, Z., Zhou, G., Hao, S., Wu, J., Gu, B.-L., Duan, W.: Adsorption of gas molecules on graphene nanoribbons and its implication for nanoscale molecule sensor. J. Phys. Chem. C 112, 13442–13446 (2008)
Huang, Z., Peng, X., Yang, H., He, C., Xue, L., Hao, G., Zhang, C., Liu, W., Qi, X., Zhong, J.: The structural, electronic and magnetic properties of bi-layered MoS2 with transition-metals doped in the interlayer. RSC Adv. 3, 12939 (2013)
Joensen, P., Crozier, E.D., Alberding, N., Frindt, R.F.: A study of single-layer and restacked MoS2 by X-ray diffraction and X-ray absorption spectroscopy. J. Phys. C 20, 4043 (1987)
Kang, J., Kim, Y.-H., Glatzmaier, G.C., Wei, S.-H.: Origin of anomalous strain effects on the molecular adsorption on boron-doped graphene. J. Chem. Phys. 139, 044709 (2013)
Kemp, K.C., Seema, H., Saleh, M., Le, N.H., Mahesh, K., Chandraa, V., Kim, K.S.: Environmental applications using graphene composites: water remediation and gas adsorption. Nanoscale 5, 3149–3171 (2013)
Kohn, W., Sham, L.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965)
Koklj, A.: Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Comput. Mater. Sci. 28, 155–168 (2003)
Kresse, G., Joubert, D.: From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999)
Late, D.J., Huang, Y.-K., Liu, B., Acharya, J., Shirodkar, S.N., Luo, J., Yan, A., Charles, D., Waghmare, U.V., Dravid, V.P., Rao, C.N.R.: Sensing behavior of atomically thin-layered MoS2 transistors. ACS Nano 7, 4879 (2013a)
Late, D.J., Huang, Y.-K., Liu, B., Acharya, J., Shirodkar, S.N., Luo, J., Yan, A., Charles, D., Waghmare, U.V., Dravid, V.P., Rao, C.N.R.: Sensing behavior of atomically thin-layered MoS2 transistors. ACS Nano 7, 4879–4891 (2013b)
Le, D., Rawal, T.B., Rahman, T.S.: Single-layer MoS2 with sulfur vacancies: structure and catalytic application. J. Phys. Chem. C 118, 5346–5351 (2014)
Lee, K., Gatensby, R., McEvoy, N., Hallam, T., Duesberg, G.S.: High-performance sensors based on molybdenum disulfide thin films. Adv. Mater. 25, 6699–6702 (2013)
Leenaerts, O., Partoens, B., Peeters, F.M.: Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B 77, 125416 (2008)
Li, H., Yin, Z., He, Q., Li, H., Huang, X., Lu, G., Fam, D.W.H., Tok, A.I.Y., Zhang, Q., Zhang, H.: Fabrication of single-and multilayer MoS2 film-based field-effect transistors for sensing NO at room temperature. Small 8, 63 (2012)
Luo, H., Cao, Y., Zhou, J., Feng, J., Cao, J., Guo, H.: Adsorption of NO2, NH3 on monolayer MoS2 doped with Al, Si, and P: A first-principles study. Chem. Phys. Lett. 643, 27–33 (2016a)
Luo, H., Cao, Y., Zhou, J., Feng, J., Cao, J., Guo, H.: Adsorption of NO2, NH3 on monolayer MoS2 doped with Al, Si, and P: a first-principles study. Chem. Phys. Lett. 643, 27–33 (2016b)
Ma, D., Ju, W., Li, T., Zhang, X., He, C., Ma, B., Lu, Z., Yang, Z.: The adsorption of CO and NO on the MoS2 monolayer doped with Au, Pt, Pd, or Ni: A first-principles study. Appl. Surf. Sci. 383, 98–105 (2016)
Mak, K.F., Lee, C., Hone, J., Shan, J., Heinz, T.F.: Atomically thin MoS2: a new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010)
Momma, K., Izumi, F.: VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011)
Monkhorst, H.J., Pack, J.D.: Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976)
Niu, F., Liu, J.-M., Tao, L.-M., Wang, W., Song, W.-G.: Nitrogen and silica co-doped graphene nanosheets for NO2 gas sensing. J. Mater. Chem. A 1, 6130–6133 (2013)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 78, 1396 (1997)
Sienicki, W., Hryniewicz, T.: Solar Energy Mater. Solar Cells 43, 67 (1996)
Soler, J.M., Artacho, E., Gale, J.D., Garca, A., Junquera, J., Ordejn, P., Snchez-Portal, D.: The SIESTA method for ab initio order-N materials simulation. J. Phys. 14, 2745–2779 (2002)
Tang, S., Cao, Z.: Adsorption of nitrogen oxides on graphene and graphene oxides: INSIGHTS from density functional calculations. J. Chem. Phys. 134, 044710 (2011)
Tang, S., Cao, Z.: Adsorption and dissociation of ammonia on graphene oxides: a first-principles study. J. Phys. Chem. C 116, 8778–8791 (2012)
Troullier, N., Martins, J.: Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993–2006 (1991)
Wang, Q.H., Kalantar-Zadeh, K., Kis, A., Coleman, J.N., Strano, M.S.: Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 7, 699–712 (2012)
Wilcoxon, J., Thurston, T., Martin, J.: Applications of metal and semiconductor nanoclusters as thermal and photo-catalysts. Nanostruct. Mater. 12, 993–997 (1999)
Yao, Y., Tolentino, L., Yang, Z., Song, X., Zhang, W., Chen, Y., Wong, C.-P.: High-concentration aqueous dispersions of MoS2. Adv. Funct. Mater. 23, 3577–3583 (2013)
Yuan, W., Shi, G.: Graphene based gas sensors. J. Mater. Chem. A 1, 10078–10091 (2013)
Yue, Q., Shao, Z., Chang, S., Li, J.: Adsorption of gas molecules on monolayer MoS2 and effect of applied electric field. Nanoscale Res. Lett. 8, 425 (2013)
Zhang, D.Z., Jiang, C.X., Li, P., Sun, Y.: Layer-by-layer self-assembly of Co3O4 nanorod-decorated MoS2nanosheet-based nanocomposite toward high-performance ammonia detection. ACS Appl. Mater. Int. 9, 6462–6471 (2017)
Zhao, S., Xue, J., Kang, W.: Gas adsorption on MoS2 monolayer from first-principles calculations. Chem. Phys. Lett. 595, 35 (2014)
Zhou, Y.G., Zu, X.T., Gao, F., Nie, J.L., Xiao, H.Y.: Adsorption of hydrogen on boron-doped graphene: a first-principles prediction. J. Appl. Phys. 105, 014309 (2009)
Acknowledgements
This work has been supported by Azarbaijan Shahid Madani University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abbasi, A., Abdelrasoul, A. & Sardroodi, J.J. Adsorption of CO and NO molecules on Al, P and Si embedded MoS2 nanosheets investigated by DFT calculations. Adsorption 25, 1001–1017 (2019). https://doi.org/10.1007/s10450-019-00121-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-019-00121-6