
Abstract
A density function theory (DFT) is applied to investigate the interaction and adsorption of nitrate ion on the exterior and interior surface of the pristine, Al and Ga-doped BNNTs. The calculated results indicate that the values of adsorption energy and enthalpy of the NO3−@ Al-doped BNNTs complex is more negative than pristine and Ga-doped. The adsorption energy nitrate ion on the surface of BNNTs is in order Al-doped > Ga-doped > pristine. This result demonstrates that the adsorption of nitrate ion on the surface of Al-doped BNNTs is stronger than Ga doped and pristine states. The chemical potential (µ) values for nitrate ion adsorption on the pristine, Al and Ga doped BNNTs are negative and is in order µpristine > µAl-doped > µGa-doped, it means that these compounds are stable. The values of ▽2ρ(BCP) and H(BCP) for [(NO3)O…B(BNNTs)] at the all adsorption models are positive and the |V/G| ratio for all models is > 2, it denotes the strong electrostatic interaction between nitrate ion with nanotube. In addition, the results of natural bonding orbital (NBO) and maximum charge transfer parameters (∆N) indicate that at all adsorption models, the charge transfer occurs from nitrate ion toward nanotube and nanotube acts as p-type semiconductor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
One of the most contaminants and pollutants compound of the environmental and surface or ground waters is nitrate ion (NO3−) (Öztürk and Bektas 2004; Xing et al. 2011). The main sources of nitrate ion in the water and environmental are industrial wastes, detergent manufacturing, chemical and natural fertilizers and municipal wastewater (Samatya et al. 2006a, b; Milmile et al. 2011). When nitrate ion is abundantly introduced into the water ways, it causes rapid growth of the liver and heavy algal and thus causes stimuli eutrophication and pollution of the environment (Conley et al. 2009; Mishra and Patel 2009). A high concentration of nitrate ions in drinking water and environmental system is a serious hazard to human health causing abnormalities such as vomiting, diarrhea, increased infant mortality, hypertension, central nervous system birth defects, respiratory tract infections diabetes, changes to the immune system and cancerous growths in the human digestion system (Mossa Hosseini et al. 2011; Fewtrell 2004; Samatya et al. 2006). Nitrate ion is a very stable, highly soluble ion in water that is difficult to remove by conventional water treatment methods such as coagulation, electrochemistry, flocculation, lime softening or surface adsorption processes (Milmile et al. 2011; Conley et al. 2009; Della Rocca et al. 2007; Mishra and Patel 2009; Mossa Hosseini et al. 2011; Hendricks 2006). In the recent years, various forms of carbon nano particle, graphene, their composites, and other compounds have been used to improve the adsorption of nitrate ions (Ganesan et al. 2013; Dai et al. 2009; Wehling et al. 2008; Chang et al. 2001; Yim et al. 2003).
One of the novel nano compounds that is most considered by empirical and theoretical researchers is boron nitride nanotube. Boron nitride nanotube (BNNTs) was first theoretically predicted in 1994 (Rubio et al. 1994) and then experimentally synthesized in 1995 by Chopra and co-workers (1995).
In the recent years, the adsorption and interaction of CO (Tontapha et al. 2013; Xie et al. 2012; Wang et al. 2014; Wang and Zhang 2008; Roohi et al. 2017), NO (Xie et al. 2012; Wang et al. 2014; Roohi et al. 2017), CO2 (Paura et al. 2014; Mahdavifar and Abbasi 2014; Shao et al. 2013; Tabtimsai et al. 2014), NH3 (Soltani et al. 2012), H2O2 (Soltani et al. 2013), Imidazole (Ahmadi Peyghan et al. 2013), Phosgene gas (Hosseinian et al. 2018), Noble gases (Wang and Guo 2017), Anti-cancer drug (Wang et al. 2017; Hesabi and Behjatmanesh-Ardakani 2017; Noei et al. 2017), Ethyl benzene (Noei et al. 2017), SO2 (Noei 2017), Chitosan (Rodriguez Juarez et al. 2013), Deuterium (Koswattage et al. 2011) Ammonia borane (Ahmadi Peyghan et al. 2014), and Nitramide (Kakemam and Noei 2014) on the surface of pristine and doped BNNTs was investigated by using theoretical methods. The results of these studies proved that pure and substituted boron nitride nanotubes with different elements can be a good absorbent for the above-mentioned pollutants. The electrical and optical properties of the BNNTs are significantly altered by the absorption of pollutant gases, which is used to provide nanosensors.
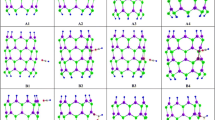
Following our pervious study (Rezaei-Sameti and Yaghoobi 2015; Rezaei-Sameti and; Moradi 2017; Rezaei-Sameti and Samadi Jamil 2016), in this work, we have decided to investigate nitrate ion adsorption on the surface of pristine, Al and Ga doped boron nitride nanotubes by using density function theory. For this purpose, we consider various different configurations for adsorption of nitrate ion on the surface of nanotube. At first step the considered models are optimized at B3LYP/3-21G level of theory, and then among of all optimized configurations, we selected the stable adsorption models without imaginary frequency. The letters of D, E and F denote to the pristine, Al and Ga-doped (8, 0) zigzag BNNTs, (a) and (e) indexes are applied to determine the adsorption of NO3− from O atoms head on the exterior surface and interior surface of nanotube. The optimized adsorption models are shown in Fig. 1. The electrical properties, thermodynamic, quantum, natural bond orbital (NBO), reduced density gradient (RDG), atom in molecule (AIM) and electrostatic potentials (ESP) are calculated and results are analyzed. The results of this study may be useful for making an adsorbent for nitrate ion.

2D views for NO3‒ adsorption on the surface of BNNTs in models D-a, D-e, E-a, E-e, F-a and F-e
2 Computational methods
In the current work, the structures of all selected adsorption models are optimized by DFT methods at the Cam-B3LYP/6-31G (d) level of theory (Zhang et al. 2014) with performing the GAMESS suite of programs (Ditchfield et al. 1972; Schmidt et al. 1993). The optimization criteria (Max. force = 0.00052, RMS force = 0.00042, Max displacement = 0.0002 and RMS displacement = 0.0008) and confirmed by vibrational frequency calculations. From optimized structures the adsorption and thermodynamic parameters of system are calculated by Eqs. (1) and (2).
The \({E_{BNNTs/{\text{NO}}_{{_{3}}}^{ - }}}\), \({E_{BNNTs}}\) and \({E_{{\text{NO}}_{{_{3}}}^{ - }}}\) obtained from potential energy of the BNNTs/NO3− complex, BNNTs and NO3− ion respectively and the BSSE is base set superposition error. The calculated results indicate that the BSSE for all adsorption models is in range 0.00065–0.00115 eV. The \({F_{BNNTs/{\text{N}}{{\text{O}}_3}}}\), \({F_{BNNTs}}\) and \({F_{{\text{N}}{{\text{O}}_3}}}\) are thermodynamic parameters such as enthalpy energy (H), Gibbs free energy (G) and entropy (S) for the BNNTs/NO3− complex, BNNTs and NO3− ion respectively. By using the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy, the gap energy (\({E_{gap}}={E_{LUMO}} - {E_{HOMO}}\)), electronic chemical potential \(\left( {\mu =\frac{{{E_{LUMO}}+{E_{HOMO}}}}{2}} \right)\), global hardness \(\left( {\eta =\frac{{{E_{LUMO}} - {E_{HOMO}}}}{2}} \right)\), and maximum charge transfer parameters \(\left( {\Delta N=\frac{{ - \mu }}{\eta }} \right)\) are calculated for all selected models (Rezaei-Sameti and Yaghoobi 2015; Rezaei-Sameti and; Moradi 2017; Rezaei-Sameti and Samadi Jamil 2016).
3 Results and discussion
3.1 The adsorption energy and thermodynamic parameters
To find the stable adsorption configuration as a preliminary test, we consider various possible adsorption geometries for nitrate ion on the exterior and interior surface of (8,0) zigzag BNNTs, and after optimizing all of the studied structures, we selected the most stable adsorption models such as D-a, D-e, E-a, E-e, F-a and F-e (see Fig. 1). The B–N bond length and gap energy of pristine (8,0) zigzag BNNTs is about 1.45 Å and 5.70 eV, which is in agreement with previous report (Jamshid 2017; Deng et al. 2016), and the average diameter of nanotube is about 6.40 Å. We can see (in Table 1), in the D-a, E-a, and F-a models the distance between nitrate ion and nanotube are as follows: 1.57, 1.85 and 1.93 Å respectively. An interesting point is that the replacement of Al and Ga atoms instead of the boron atom (in the E-a and F-a models) increases the bonding distance between the nanotubes and the nitrate ion. According to the calculated results of Table 1, with doping of Al and Ga atoms (in the E-a and F-a models), the amount of adsorption energy and enthalpy of system are become more negative than pristine models. The negative values of adsorption and enthalpy energies of system indicate an attractive interaction between the nitrate ion and BNNTs; thus, it also indicates the combining processes of the hybrid systems are exothermic. It is interesting to note that by doping Al atom (E-a model), the adsorption process is more favorable than the replacement of Ga atom (F-a model). The more negative values of absorption and enthalpy energies show that the nitrate ion has strong chemical interaction with the Al-doped BNNTs, thus the stable adsorbed structures are formed. Inspection of results indicate that with doping Al and Ga atoms the adsorption of nitrate ion on the exterior and interior layer of nanotube increase significantly from pristine model, the increment of adsorption nitrate ion on the exterior and interior surface of nanotubes is in order Al-doped(E-a) > Ga-doped(F-a) > pristine(D-a) and Ga-doped(F-e) > Al-doped(E-e) > pristine(D-e) respectively. The values of dipole moment indicate that the polarity of system decreases by doping Al&Ga atoms and adsorbing of nitrate ion on the surface of nanotube, which is the desired attribute for nitrate ion adsorption from environmental. The calculated results demonstrate that the adsorption energy and enthalpy of the adsorption process have inverse trend with the dipole moment. In addition the results of NBO and Mulliken charges show that upon adsorption of nitrate ion on the exterior and interior surface of pristine, Al and Ga doped BNNTs, the charge transfer occurs from nitrate ion toward nanotube and nanotube acts as p-type semiconductor. The Gibbs free energy of NO3−@ nanotube complex is negative and indicates that the adsorption process is spontaneously in view of thermodynamic approach.
3.2 Electronic structure properties of system
To understand the electronic properties of NO3− adsorption on the exterior and interior surface of pristine, Al and Ga doped BNNTs we calculated the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) and quantum parameters. The HOMO and LUMO plots are shown in Fig. 2 and the calculated quantum parameters such as HOMO and LUMO energy; Fermi level energy, gap energy (Egap), global hardness (η), electro chemical potential (µ) and charge transfer parameters (∆N) are presented in Table 2. As we know HOMO orbital is the outer orbital containing electrons, tend to give these electrons as an electron donor and hence the ionization potential is directly related to the energy of the HOMO and LUMO orbital accept electrons and the LUMO energy is directly related to electron affinity. According to calculated results of Fig. 2, the HOMO orbital density at the D-a, E-a and F-a models are localized on the surface of nanotube around adsorption position, while the LUMO orbital density is localized on surface of nanotube at the backside of adsorption position. On the other hand, when nitrate ion is adsorbed to the inside of nanotube, the HOMO orbital density is distributed over the surface of the nanotube, and the LUMO orbitals are concentrated on the nitrate ion. Therefore, in the process of adsorption of nitrate ion, the electron charge is transferred from the nitrate ion toward the nanotube surface, and this result is in agreement with positive values charge transfer parameters (∆N). The positive values of ∆N demonstrate that the nitrate ion has donor electron effect and is a donor electron. Comparison results indicate that with doping Al and Ga atoms, the ∆N values increase and so the donor electron effects of nitrate ion increase.

Plots of HOMO and LUMO orbital structures for NO3‒ adsorption on the surface of BNNTs in models D-a, D-e, E-a, E-e, F-a and F-e
The chemical potential (µ) of the pristine (D), Al-doped (E), Ga-doped (F) and nitrate ion adsorptions models are negative and it means that these compounds are stable. They do not decompose spontaneously into the elements and compounds are made up of them. Comparison results display that with doping Al and Ga atoms the chemical potential of system decrease from pristine model and is in order µD > µE > µF. The global hardness shows the resistance towards the deformation of electron cloud of system under small perturbation encountered during the chemical process. The global hardness of BNNTs with doping Al and Ga atom and absorbing nitrate ion reduce from original state and the activity of system increase. One of the important parameters to study the electrical and reactivity properties of system is gap energy (Egap). The Egap of the pristine, Al-doped, and Ga-doped BNNTs is 6.38, 7.85 and 7.53 eV respectively. The gap energy of the E-a, E-e and F-a models is more than unabsorbed nanotube, whereas the gap energy of the D-a, D-e and F-f models is more than unabsorbed nanotube. As we know the global hardness and gap energy are related directly to reactivity and conductivity of system, i.e. the molecule with the least gap energy is little hardness and more reactive and conductive. As a result, the activity and conductivity of the D-a, D-e and F-f models is lower than pristine state.
The densities of state (DOS) plots for in title adsorption models are determined by using GaussSum program (O’Boyle et al. 2008) in energy range of − 15 to + 10 eV and results are shown in Fig. 3. By using DOS plots we can find the number of states per interval of energy at each level of energy that are available to be occupied by electrons. A high DOS at a certain level of energy means that there are many states available for occupation. Inspection of DOS plots indicate that the number of peaks at the HOMO region in the D-a, E-a and F-a models are 11 and for D-e, E-e and F-e models are 9 peaks. On the LUMO region the number of peaks in the D-a, E-e, F-e models are 10 and for E-a, F-a and D-e models are 8 peaks. On the other hand, the valence and conduction levels of nanotube for adsorption of nitrate ion on the exterior and interior surface of BNNTs slightly shift upward, which is negligible, and close to that of the pristine nanotube.

DOS Plots for NO3‒ adsorption on the surface of BNNTs in models D-a, D-e, E-a, E-e, F-a and F-e
3.3 Natural bond orbital (NBO) analysis
The natural bond orbital method is used for exploring charge transfer in molecular interaction and intra- molecular bonding between two compounds. The NBO analysis is an effective tool for the chemical and electron density transfer from the filled lone pair electron. In this work for each donor (i) and acceptor (j) the stabilization energy (E(2)) associated with the delocalization i → j is determined as (Bulat et al. 2010):
where qi is donor orbital occupancy, \({\varepsilon _i}\,\,\) and \({\varepsilon _j}\,\,\) are orbital energies and Fij is the off-diagonal NBO Fock matrix element. The larger of E(2) value reveals the more intensive interaction between electron donors and electron acceptors, i.e. the more donating tendency from electron donors to electron acceptors of the whole system. The possible intensive interactions between nitrate ion and pristine, Al and Ga doped BNNTs are listed in Table 3. The second-order perturbation theory analysis of Fock matrix in NBO basis shows the strong intramolecular interactions occur between σB62-N52 → σ*N52-B63. The obtained E(2) values for interaction between above studied orbital for D-a, D-e, E-a, E-e, F-a and F-e models are 6.97, 6.94, 5.86, 5.14, 6.01 and 5.01 Kcal/mol respectively. Comparison results indicate that the E(2) values for adsorbing nitrate ion on the outer surface of pristine, Al and Ga doped BNNTs is more than inner surface. It is noteworthy that with doping Al and Ga atom the E(2) values decrease significantly from pristine model and so the intramolecular interaction between doped nanotube and nitrate ion is lower than pristine model. The weak intramolecular interactions occur between σN32-B22 → σ*N51-B41. The obtained E(2) values for interaction between above studied orbital in D-a, D-e, E-a, E-e, F-a and F-e models are 2.08, 3.47, 3.97, 2.98, 3.60 and 2.99 Kcal/mol respectively.
On the other hand, for interaction between σN52-B36 → σ*N51-B62 (donor–acceptor orbital) the stabilization energy for D-a, D-e, E-a, E-e, F-a and F-e models are 5.23, 4.10, 4.45, 3.97, 3.93 and 3.29 Kcal/mol respectively. It is found that the stabilization energy of pristine model is more than Al and Ga-doped models.
3.4 Molecular electrostatic potential (MEP)
Molecular electrostatic potential (MEP) is one the most important and useful method to predict the best site of interaction for the donor–acceptor system, from which the electrophilic and nucleophilic positions. In the MEP plots, the most positive electrostatic potential (blue color) is the highest electrophilic site, and the negative regions of electrostatic potential (red color) which are nucleophilic sites are associated with the lone pair of electronegative atoms.
The calculated MEP plots for adsorption position of nitrate ion with nanotube are shown in Fig. 4. As it is seen the positive sites for D-a, E-a, F-a, D-e, E-e and F-e models are located on the nitrate ion and adsorption position and the negative sites are located on surface of nanotube. Which shows charge transfer takes place between nitrate ion and nanotube and that sites of nitrate ion and adsorption position are suitable for the adsorption process.

The MEP Plots for NO3‒ adsorption on the surface of BNNTs in models D-a, D-e, E-a, E-e, F-a and F-e
3.5 QTAIM topological analysis
The theory of atoms in molecules was introduced for the first time by Bader (1990). This theory defines the chemical structure of a system as based on the distribution of electron density between two atoms. Bader described four types of critical points, one of them being a saddle point of electron density between two atoms forming a chemical bond is called bond critical point (BCP). Besides the charge density in BCP, its Laplacian (representing the local charge concentration or depletion) and ellipticity (the measure of “double bond” or π character) are also useful parameters. The Laplacian parameters at the BCP is given by \({\nabla ^2}\rho ={\lambda _1}+{\lambda _2}+{\lambda _3}\), where λ1, λ2 and λ3 are three parameters of the density at the critical point. The electronic energy density (H) is the sum of kinetic energy density (G), potential energy density (H(BCP) = G(BCP) + V(BCP)) and helps more accurate descriptions of the nature of bonds. Positive ▽2ρ and H values denote the weak covalent interactions (strong electrostatic bond), negative ▽2ρ and H values refer to strong interaction (strong covalent bond), and medium strength (▽2ρ > 0 and H < 0) is defined as partially covalent bond. On the other hand, the |V/G| ratio is a reliable parameter to classify the different interactions. Based on this parameter, weak interactions are associated with |V/G| < 1, medium interactions 1 < |V/G| < 2, and strong interactions |V/G| > 2. The values of electron density (ρ), Laplacian (▽2ρ), kinetic energy density (G), potential energy density (V), total energy density (H), and the ratio of |V/G| for interaction O atom of nitrate ion with B atom of nanotube [(NO3)O…B (BNNTs)] for the D-a, E-a, F-a, D-e, E-e and F-e models are listed in Table 4. The BCP plots for all adsorption models are shown in Fig. 5. Inspection of results demonstrate that the values of ▽2ρ and H(BCP) for [(NO3)O…B (BNNTs)] for all adsorption models are positive denote electrostatic interaction and the |V/G| ratio for all models is > 2 denote strong interaction. The |V/G| ratio for E and F models (Al and Ga doped BNNTs) is more than pristine BNNTs and so the interaction and adsorption of nitrate ion with Al and Ga doped is stronger than pristine model. This result confirms that Al and Ga doped BNNTs is a good adsorbent to nitrate ion. The large values of the ellipticity ε (> 0.1) correspond to double bonds and that lower values correspond to broken double bonds or the formation of single bonds. In this study it is found that the ε values for D-e and E-a models are 0.1777 and 0.1623 eV and for other models is lower than 0.1 eV. According to the ellipticity (ε) values, the bonding between nitrate ion and nanotube is single bond type.

The bond critical position for NO3‒ adsorption on the surface of BNNTs in models D-a, D-e, E-a, E-e, F-a and F-e
3.6 Reduced density gradient (RDG) and NCI index
To determine the intramolecular interactions and evaluate the nature of the weak interactions, the non-covalent interaction index (NCI) for the complexes considered have been calculated. The NCI index provides more evidence related to the non-covalent interaction. The reduced density gradient (RDG) is defined (Johnson et al. 2010):
Non-covalent interactions are characterized by small values of RDG. These isosurface expand over interacting regions of the complex. The product between electron density ρ(r) and the sign of the second lowest eigenvalues of electron density hessian matrix (λ2) has been proposed as a tool to distinguish the different types of interactions. The scatter graphs of RDG versus sign(λ2)ρ(r) for all adsorption models are shown in Fig. 5. The X-axis and Y axis are sign(λ2)ρ(r) and RDG function respectively. The sign(λ2)ρ(r) and NCI–RDG plots are obtained with Multiwfn program (Runge and Gross 1984). The sign (λ2) is utilized to distinguish the bonded (λ2 < 0) interactions from nonbonding (λ2 > 0) interactions. In the RDG scatter graph blue color circle shows the attractive interactions, red color circle denotes strong repulsive interactions and green circle implies low electron density, corresponding to Van der walls interactions. These isosurface are located on the reaction sites of the NO3−/BNNTs complex. It clearly observed that with doping Al and Ga atom more electron density localized in λ2 < 0 region and the attractive interactions is increase. It is notable that the density of electron in the Al doped models is more than Ga doped and so Al doped is more suitable than Ga doped for nitrate ion adsorption. On the other the attraction and repulsion interaction in the inner layer of nanotube (Al and Ga doped BNNTs or E-e and F-e models) is more obvious than other models. Therefore, the adsorption of nitrate ion in the inner layer and presence of Al and Ga doped BNNTs is more favorable than pristine model (Fig. 6).

The RDG/sign(λ2)ρ for NO3‒ adsorption on the surface of BNNTs in models D-a, D-e, E-a, E-e, F-a and F-e
4 Conclusions
In this work, the effects of Al and Ga doped atoms on the interaction of nitrate ion on the exterior and interior surface of BNNTs is investigated by using computational methods. The calculated results demonstrate that adsorption of nitrate ion on the exterior and interior surface is in order Al-doped(E-a) > Ga-doped(F-a) > pristine(D-a) and Ga-doped(F-e) > Al-doped(E-e) > pristine(D-e) respectively. This result confirms that Al doped BNNTs is a good candidate to nitrate ion adsorption. The |V/G| ratio for E and F models (Al and Ga doped BNNTs) is more than pristine BNNTs and so the interaction and adsorption of nitrate ion with Al&Ga doped is stronger than pristine model. The RDG plots show that with doping Al and Ga atom more electron density localized in λ2 < 0 region and the attractive interactions is increase. The MEP results indicate that the charge transfer occur from nitrate ion toward nanotube.
References
Ahmadi Peyghan, A., Baei, M.T., Moghimi, M., Hashemian, S.: Adsorption and electronic structure study of imidazole on (6,0) zigzag single-walled boron nitride nanotube. J. Clust. Sci. 24, 31–47 (2013)
Ahmadi Peyghan, A., Aslanzadeh, S.A., Samiei, A.: Ammonia borane reaction with a BN nanotube: a hydrogen storage route. Monatsh. Chem. 145, 1083–1087 (2014)
Bader, R.F.W.: Atoms in Molecules: A Quantum Theory. Oxford University Press, Oxford (1990)
Bulat, F.A., Toro-Labbé, A., Brinck, T., Murray, J.S., Politzer, P.: Quantitative analysis of molecular surfaces: areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 16(11), 1679–1691 (2010)
Chang, H., Lee, J.D., Lee, S.M., Lee, Y.H.: Adsorption of NH3 and NO2 molecules on carbon nanotubes. Appl. Phys. Lett. 79, 3863–3865 (2001)
Chopra, N.G., Luyken, R.J., Cherrey, K., Crespi, V.H., Cohen, M.L., Louie, S.G., et al.: Boron nitride nanotubes. Science 269, 966–967 (1995)
Conley, D.J., Paerl, H.W., Howarth, R.W., Boesch, D.F., Seitzinger, S.P., Havens, K.E., Lancelot, C., Likens, G.E.: Controlling eutrophication: nitrogen and phosphorus. Science 323, 1014–1015 (2009)
Dai, J., Giannozzi, P., Yuan, J.: Adsorption of pairs of NOx molecules on single-walled carbon nanotubes and formation of NO + NO3 from NO2. Surface Sci. 603, 3234–3238 (2009)
Della Rocca, C., Belgiorno, V., Meriç, S.: Overview of in-situ applicable nitrate removal processes. Desalination 204, 46–62 (2007)
Deng, Z.-Y., Zhang, J.-M., Xu, K.-W.: Adsorption of SO2 molecule on doped (8, 0) boron nitride nanotube: a first-principles study. Physica E 76, 47–51 (2016)
Ditchfield, R., Hehre, W.J., Pople, J.A.: Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724–728 (1972)
Fewtrell, L.: Drinking-water nitrate, methemoglobinemia, and global burden of disease: a discussion. Environ. Health Perspect. 112, 1371–1374 (2004)
Ganesan, P., Kamaraj, R., Vasudevan, S.: Application of isotherm, kinetic and thermodynamic models for the adsorption of nitrate ions on graphene from aqueous solution. J. Taiwan Inst. Chem. Eng. 44, 808–814 (2013)
Hendricks, D., Water Treatment Unit Processes. Taylor and Francis Group, Boca Raton (2006)
Hesabi, M., Behjatmanesh-Ardakani, R.: Interaction between anti-cancer drug hydroxycarbamide and boron nitride nanotube: a long-range corrected DFT study. Comput. Theo. Chem. 1117, 61–80 (2017)
Hosseinian, A., Salary, M., Arshadi, S., Vessally, E.: The interaction of phosgene gas with different BN nanocones: DFT studies. Solid State Commun. 269, 23–27 (2018)
Jamshid, N.: Modulating band gap and HOCO/LUCO energy of boron-nitride nanotubes under a uniform external electric fiel. Iran. J. Chem. Chem. Eng. 36, 93–106 (2017)
Johnson, E.R., Keinan, S., Mori-Sanchez, P., Contreras-Garcia, J., Cohen, A.J., Yang, W.: Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 (2010)
Kakemam, J., Noei, M.: Density functional study on the functionalization of BN nanotubes with nitramide. Russ. J. Phys. Chem. A. 88, 1751–1756 (2014)
Koswattage, K.R., Shimoyama, I., Baba, Y., Sekiguchi, T., Nakagaw, K.: ab Study on selective adsorption of deuterium on boron nitride using photon-stimulated ion-desorption. App. Surface Sci. 258, 1561–1564 (2011)
Mahdavifar, Z., Abbasi, N.: The influence of Cu-doping on aluminum nitride, silicon carbide and boron nitride nanotubes’ ability to detect carbon dioxide; DFT study. Physica E 56, 268–276 (2014)
Milmile, S.N., Pande, J.V., Karmakar, S., Bansiwal, A., Chakrabarti, T., Biniwale, R.B.: Equilibrium isotherm and kinetic modeling of the adsorption of nitrates by anion exchange Indion NSSR resin. Desalination 276, 38–44 (2011)
Mishra, P.C., Patel, R.K.: Use of agricultural waste for the removal of nitrate-nitrogen from aqueous medium. J. Environ. Manag. 90, 519–522 (2009)
Mossa Hosseini, S., Ataie-Ashtiani, B., Kholghi, M.: Nitrate reduction by nano-Fe/Cu particles in packed column. Desalination 276, 214–221 (2011)
Noei, M.: Different electronic sensitivity of BN and AlN nanoclusters to SO2 gas: DFT studies. Vacuum. 135, 44–49 (2017)
Noei, M., Ahmadaghaei, N., Salari, A.: A., Ethyl benzene detection by BN nanotube: DFT studies. J. Saudi Chem. Soc. 21, S12–S16 (2017)
O’Boyle, N., Tenderholt, A., Langner, K.: A library for package-independent computational chemistry algorithms. J. Comp. Chem. 29, 839–845 (2008)
Öztürk, N., Bektas, T.E.: Nitrate removal from aqueous solution by adsorption onto various materials. J. Hazard. Mater. B. 112, 155–162 (2004)
Paura, E.N.C., da Cunha, W.F., Martins, J.B.L., e Silva, G.M., Roncaratti, L.F., Gargano, R.: Carbon dioxide adsorption on doped boron nitride nanotubes. RSC Adv. 4, 28249–28258 (2014)
Rezaei-Sameti, M., Samadi Jamil, E.: The adsorption of CO molecule on pristine, As, B, BAs doped (4,4) armchair AlNNTs: a computational study. J. Nanostruct. Chem. 3, 1–9 (2016)
Rezaei-Sameti, M., Yaghoobi, S.: Theoretical study of adsorption of CO gas on pristine and AsGa-doped (4, 4) armchair models of BPNTs. Comput. Condens. Matter 3, 21–29 (2015)
Rezaei–Sameti, M., Moradi, F.: Interaction of isoniazid drug with the pristine and Ni-doped of (4, 4) armchair GaNNTs: a first principle study. J. Incl. Phenom. Macrocycl. Chem. 88, 209–218 (2017)
Rodriguez Juarez, A., Chigo Anota, E., Cocoletzi, H., Flores Riveros, A.: Adsorption of chitosan on BN nanotubes: a DFT investigation. App. Surface Sci. 268, 259–264 (2013)
Roohi, H., Maleki, L., Erfani Moradzadeh, M.: Exploring electronic properties and NO gas sensitivity of Si-doped SW-BNNTs under axial tensile strain. J. Mater. Sci. 52, 9739–9763 (2017)
Rubio, A., Corkill, J.L., Cohen, M.L.: Theory of graphitic boron nitride nanotubes. Phys. Rev. B 49, 5081 (1994)
Runge, E., Gross, E.K.U.: Density-functional theory for time-dependent systems. Phy. Rev. Lett. 52, 997–1000 (1984)
Samatya, S., Kabay, N., Yuksel, U., Arda, M., Yuksel, M.: Removal of nitrate fromaqueous solution by nitrate selective ion exchange resins. React. Funct. Polym. 66, 1206–1214 (2006a)
Samatya, S., Kabay, N., Yuksel, U., Arda, M., Yuksel, M.: Removal of nitrate from aqueous solution by nitrate selective ion exchange resins. React. Funct. Polym. 66, 1206–1214 (2006b)
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M., Montgomery, J.A.: General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993)
Shao, P., Kuang, X.Y., Ding, L.P., Yang, J., Zhong, M.M.: Can CO2 molecule adsorb effectively on Al-doped boron nitride single walled nanotube? Appl. Surf. Sci. 285, 350–356 (2013)
Soltani, A., Raz, S.G., Rezaei, V.J., Khalaji, A.D., Savar, M.: Ab initio investigation of Al-and Ga-doped single-walled boron nitride nanotubes as ammonia sensor. Appl. Surf. Sci. 263, 619–625 (2012)
Soltani, A., Ahmadi Peyghan, A., Bagheri, Z.: H2O2 adsorption on the BN and SiC nanotubes: a DFT study. Physica E 48, 176–180 (2013)
Tabtimsai, C., Nonsri, A., Gratoo, N., Massiri, N., Suvanvapee, P., Wanno, B.: Carbon monoxide adsorption on carbon atom doped perfect and Stone–Wales defect single-walled boron nitride nanotubes: a DFT investigation. Monatsh Chem. 145, 725–735(2014)
Tontapha, S., Ruangpornvisuti, V., Wanno, B.: Density functional investigation of CO adsorption on Ni-doped single-walled armchair (5, 5) boron nitride nanotubes. J. Mol. Model. 19, 239–245 (2013)
Wang, C., Guo, C.: The noble gases adsorption on boron-rich boron nitride nanotubes: a theoretical investigation. Superlat. Microstr. 107, 97–103 (2017)
Wang, R.X., Zhang, D.J.: Theoretical study of the adsorption of carbon monoxide on pristine and silicon-doped boron nitride nanotubes. Aust. J. Chem. 61, 941–945 (2008)
Wang, R.X., Zhang, D.J., Liu, C.B.: The germanium-doped boron nitride nanotube serving as a potential resource for the detection of carbon monoxide and nitric oxide. Comput. Mater. Sci. 82, 361–366 (2014)
Wang, R., Zhang, D., Liu, C.: DFT study of the adsorption of 2,3,7,8-tetrachlorodibenzo-p-dioxin on pristine and Ni-doped boron nitride nanotubes. Chemosphere 168, 18–24 (2017)
Wehling, T.O., Noveselov, K.S., Morozov, S.V., Vdovin, E.E., Katsnelson, M.I., Geim, A.K., Lichtenstein, A.I.: Molecular doping of graphene. Nano Lett. 8, 173–177 (2008)
Xie, Y., Huo, Y.P., Zhang, J.M.: First-principles study of CO and NO adsorption on transition metals doped (8, 0) boron nitride nanotube. Appl. Surf. Sci. 258, 6391–6397 (2012)
Xing, X., Gao, B.-Y., Zhong, Q.-Q., Yue, Q.-Y., Li, Q.: Sorption of nitrate onto amine-crosslinked wheat straw: characteristics, column sorption and desorption properties. J. Hazard. Mater. 186, 206–211 (2011)
Yim, W.L., Gong, X.G., Liu, Z.F.: Chemisorption of NO2 on carbon nanotubes. J. Phys. Chem. B. 107, 9363–9369 (2003)
Zhang, M.L., Ning, T., Zhang, S.Y., Li, Z.M., Yuan, Z.H., Cao, Q.X.: Response time and mechanism of Pd modified TiO2 gas sensor. Mater. Sci. Semicond. Process. 17, 149–154 (2014)
Acknowledgements
The author thanks the Computational information center of Malayer University for providing the necessary facilities to carry out the research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rezaei-Sameti, M., Zarei, P. NBO, AIM, HOMO–LUMO and thermodynamic investigation of the nitrate ion adsorption on the surface of pristine, Al and Ga doped BNNTs: A DFT study. Adsorption 24, 757–767 (2018). https://doi.org/10.1007/s10450-018-9977-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-018-9977-7