
Abstract
Wall shear stress (WSS) is an important stimulus in vascular remodelling and vascular lesion development. The current methods to assess and predict the risk associated with specific unsteady WSS consider the WSS mean values or the presence of reverse phases described by the oscillatory shear index. Recent evidence has shown that the accuracy of these methods is limited, especially with respect to the venous environment. Unsteady WSS are characterised by several features that may individually affect endothelial cells. Consequently, we assessed the effects of averaged WSS (TAWSS), temporal WSS gradient (TWSSG), maximum WSS (WSS peak) and reverse phase (OSI) by applying different WSS profiles to venous EC in-vitro, using a real-time controlled cone-and-plate cell-shearing device for 24 h. We found that TWSSG and WSS peak affect cell elongation and alignment respectively. We also found that the WSS waveforms with a peak of 1.5 Pa or higher significantly correlate with the induction of a protective phenotype. Cell phenotype induced by these high peak waveforms does not correlate to what is predicted by the hemodynamic indices currently used. The definition of reliable hemodynamic indices can be used to inform the computational models aimed at estimating the hemodynamic effects on vascular remodelling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The wall shear stress (WSS) is the frictional force per unit of area exerted at the interface of the flowing blood and the endothelium. WSS is a potent stimulus that is capable of evoking varying phenotypes in endothelial cells (EC).8,35 Both in-vivo and in-vitro observations have demonstrated the existence of two major pathways that may activate EC as a response to different WSS waveforms. These two pathways are involved in inducing EC quiescence conferring a protective effect on endothelium or EC activation and pro-inflammatory signalling causing endothelium dysfunction, inflammation and pro-thrombotic effects.7 Physiologically, EC quiescence participates in vascular homeostasis while EC activation can be elicited by acute or chronic change in blood flow in order to re-establish physiological perfusion.12 The extent these mechanisms are involved in the adaptations of venous tissues used as grafts in surgical procedures remains to be elucidated.
Studies focused on the arterial context and in particular on the development of atherosclerotic lesions have shown that vascular lesions are preferentially located in certain vascular geometries where curvatures or branches cause low and disturbed hemodynamic patterns.9,45 From these observations, researchers defined the arterial unidirectional, laminar WSS (> 1–1.5 Pa) as protective. On the contrary, low WSS (< 0.4 Pa) and the presence of reverse phases in which the blood changes the flow direction, have been defined as atheroprone due to their possible role in inducing atherosclerotic plaque growth and instability.30 Numerous in-vitro experiments have demonstrated that idealised steady or unsteady WSS based on these definitions are capable of evoking an alternative EC phenotype. EC exposed to different WSS waveforms, experience a different activation state that is fundamental to flow-induced vessel remodelling and vascular lesion formation which can be established within 24 h of the flow onset in-vitro.1 In particular quiescent EC exhibit high levels of Krüppel-like factors 2 and 435,42 (KLF-2 and KLF-4), two proteins involved in the cytoskeletal remodelling3 with positive effects on vasodilation, anti-inflammatory and anti-thrombotic7 responses. Conversely, activated EC show nuclear localisation of nuclear factor kB16 (NF-kB) and high expression of pro-inflammatory signals such as interleukin 8 (Il-8)10 and adhesive molecules such as vascular cell adhesion molecule 1 (VCAM-1).6
The majority of the studies focused on arterial-like conditions in terms of WSS magnitude. The mechano-biology of venous tissues has been only partially addressed. Venous conduits are commonly used for example in coronary or peripheral bypass surgery and arteriovenous fistulae, used for vascular accesses in hemodialysis patients. Post procedure, venous tissues are exposed to mechanical stimuli typical of the arterial context. The stimuli have been demonstrated to be involved in the tissue remodelling and in the abnormal growth of neo-intimal hyperplasia causing the stenosis of the vessels29 and ultimately the failure of the surgical intervention.
Studies focused on the side-to-end arteriovenous fistula (AVF), have shown extremely disturbed hemodynamic patterns at the anastomosis level and especially in the venous curvature.20,41 These patterns correlate with development of neo-intimal hyperplasia that is the primary cause of AVF failure. Ideally, after AVF creation, increased blood flow induces a chronic change in the vascular tissue leading to an outward remodelling capable of accommodating the newly established blood flow.4,5 Previous work19 demonstrated that peak flow is specifically associated to the radial artery remodelling in response to access creation. However, AVF failure due to stenosis is preferentially located in the venous side of the anastomosis and the effects of WSS peak on venous tissues have not been addressed previously. Physiological WSS waveforms, especially those derived from complex geometries exposed to development of neo-intimal hyperplasia, are extremely unsteady and characterised by velocity fluctuations at frequencies much higher than the cardiac cycle frequency. Computational studies focused on AVF have shown these frequencies may reach 100 Hz.40 This leads to further questions regarding how venous EC behave in response to pulsatile WSS waveforms and to what extent cells can react to the distinctive features of disturbed WSS patterns.
The hemodynamic indices developed for the arterial flows, such as the time-averaged WSS (TAWSS), the oscillatory shear index33 (OSI) or the temporal WSS gradient (TWSSG) should describe the vascular risk associated with physiological WSS waveforms. These indices consider only certain features of the WSS waveforms and ultimately are not accurate in predicting vascular lesion formation.37 Furthermore, venous physiological flows and venous EC mechanotransduction have not been considered in the development of these indices. The OSI index is defined as
where τw is the WSS and T the time interval or the cardiac period. TWSSG is defined as
where τw is the WSS and dt is an interval of time.
In general, venous tissues are exposed to low, quasi-steady WSS30 ranging 0.1 to 0.6 Pa in-vivo. When exposed to arterial flow, venous EC can sustain the new hemodynamic conditions and induce the vessel remodelling in order to establish a new equilibrium. Despite the high failure rate of the AVF,34 those that succeed in the maturation process and sustain hemodialysis demonstrate that it is possible for venous tissue to durably adapt to the arterial context. To what extent the theories of WSS mechanotransduction derived from arterial studies apply to the venous context, remain to be investigated. In particular, the role of the rapid transients of WSS characterized by high peak and low average on the venous EC phenotype has not been previously addressed.
To address the specified problems, the role of WSS features such as TAWSS, temporal WSS gradient (TWSSG), maximum WSS (WSS peak) and reverse phase (OSI, in the presence of a reverse phase), on EC phenotype has been investigated. The waveforms obtained have then been tested for their protective effects on venous EC in culture focusing on flow-induced morphological adaptations and both gene and protein expressions, known to be involved in vascular lesion development. The overall aim is to systematically test these features and evaluate their role in venous EC activation and signalling.
Materials and Methods
To decouple the key factors within the inherent WSS features in-vivo, ten different idealised waveforms were created (Fig. 1) to evaluate the effects of WSS magnitude, temporal WSS gradient (TWSSG), WSS peak and reverse phases on the EC in culture. Waveforms ranging in WSS magnitude from 0.5 to 2.5 Pa, TWSSG from 2 to 20 Pa s−1 and from 0 to 0.5 of oscillatory shear index (OSI) value. The sinusoidal waveforms (Sin 0.5, Sin 1.5 and Sin 2.5) have an OSI value greater then 0, a magnitude peak ranging from 0.5 to 2.5 Pa and a reverse phase with a magnitude of − 0.5 Pa. Pulsatile waveforms and their fast (F) or very fast (VF) versions (Pul 0.5, Pul 0.5 F, Pul 0.5 VF, Pul 1.5, Pul 1.5 F, Pul 2.5, Pul 2.5 F) have an OSI value of 0, a peak magnitude ranging from 0.5 to 2.5 Pa and no reverse phase. To evaluate the increase of TWSSG at the same peak, the waveforms were modified to approximately double the gradient of the initial acceleration phases that ranges from 2 to 20 Pa s−1. It is important to notice that despite a significant peak magnitude, all waveforms are characterised by a low WSS average (0 to 0.8 Pa) due to the presence of the reverse phase or of a long resting phase (0 Pa). All waveforms have a frequency of 1Hz. Waveforms characteristics are shown in the table of Fig. 1.

Wall shear stress (WSS) waveforms tested on endothelial cells (ECS) in culture. In the table are reported the waveform features of interest. Sin sinusoidal, Pul pulsatile, TAWSS time-averaged wall shear stress, OSI oscillatory shear index, TWSSG temporal wall shear stress gradient.
Pooled human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (CC-2519, Basel, Switzerland). Cells were expanded until 4th passage on plates (Falcon, 150 mm TC-treated Cell Culture Dish, Corning, Corning, USA) precoated with 1% bovine gelatine (G1393,Sigma, St. Louis, USA) using Endothelial Cell Growth Basal Medium-2 (EGM-2, Lonza) supplemented with Endothelial Cell Growth Medium-2 BulletKit (Lonza), 1% Amphotericin B (A2942, Sigma-Aldrich) and 1% penicillin-streptomycin (P4333, Sigma).
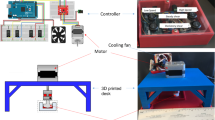
EC were exposed to different WSS stimuli for 24 h using a real-time controlled cone-and-plate device (see Supplementary information, SI). The cone is designed with an angle of 0.5° to ensure laminar flows and avoid the insurgence of secondary flows for all tested conditions.24 This allows for a controlled WSS delivered to cell in culture.
Previous studies showed that that the endothelial activation state and the expression of several genes are regulated by flow exposure within 24 h.1,28,46,47 However, the upregulation of KLF-2 and KLF-4, two fundamental genes that induce the protective (quiescent) phenotype in endothelial cells can also be detected within 24 h23,36 and to our knowledge there are no results that showed endothelial cells KLF-2 upregulation detectable only after exposure longer than 24 h. Furthermore, Small GTPases transient activation is resolved within 2 h.47 These proteins, in the presence of KLF-2, control the alignment of the cytoskeleton. This event appears crucial for the induction of the quiescent phenotype.44 KLF-2 overexpression causes downstream events that are extremely important for the regulation of the vascular tone and the paracrine signalling within 7 days.3,22
EC exposed to different flow conditions were stained for CD31 (or platelet endothelial cell adhesion molecule, PECAM-1), f-actin, NFkB-3 and KLF-2, according to manufacturer instructions (see SI). Each condition was replicated three times. Identification of morphological changes in the ECs was performed through the digital analysis of the immunofluorescence images as previously described.25 CD-31 staining images were used to evaluate the individual cell elongation, quantified as the ratio between the major and the minor axes (Fig. 3a), by using CellProfiler software (3.1.5, Broad Institute, Cambridge, MA). The F-actin alignment process was quantified by using the OrientationJ plugin (EPFL, Lausanne, Switzerland) of the image processing software ImageJ (National Institutes of Health, Bethesda, MD). Alignment of fibres was considered to be significant within ± 10° with respect to the flow direction (see SI). Data are presented as percentage of fibres aligned.
For gene expression, RT-PCR was performed as previously described18 (see SI). Selected genes were quantified using three technical replicates and each WSS condition was tested three-to-five times. Delta-delta-Ct analysis was used to determine gene expression fold change induced by WSS stimulation compared to the static control.
Post exposure to WSS stimuli or static conditions, the protein levels were obtained using Western Blot technique (see SI). Proteins levels were evaluated using a Fluorescence Western Blot analysis system (Odyssey Fc Imager, LI-COR Biosciences, Ltd., UK) and normalized over GAPDH expression levels. Each condition was tested three-to-five times.
The presented results are expressed as the mean ± standard deviation (SD). Data were evaluated using one-way ANOVA, and differences among groups were established using a post hoc Bonferroni’s multiple-comparisons test (Prism, GraphPad Software, La Jolla, CA). The significance was established for p < 0.05.
Results
HUVECs have been successfully exposed to different WSS waveforms for 24 h. The described experimental method maintained sterility and viability of the cell cultures.
As expected,15,25 we found that HUVECs morphology was diversely affected by the different WSS waveforms (Fig. 2). EC exposed to WSS show a more uniform and continuous distribution of CD-31 (Fig. 3) protein at the cell junctions in comparison to the static flow. Furthermore, ECs exposed to sinusoidal WSS profiles show an increasingly organised and tight cell junction with the increase of the magnitude peak. No major differences are observable in cells exposed to different pulsatile WSS.

Bright light microscopy images of HUVECs after 24 h of exposure to the different WSS stimuli. Each condition was tested six times. Scale bar 50 μm.

CD-31 (PECAM) and F-actin immunofluorescence images of HUVECs after 24 hrs of exposure to the different WSS stimuli. Scale bar 20 μm. Each condition was tested three times. In the table are reported the numerical values of cell elongation and fibres alignment expressed as mean ± SD. The significance was established for p < 0.05. (a) Cell elongation of HUVECs exposed to 24 h of different WSS patterns with * representing significance with respect to the Static, Sin 0.5, Sin 1.5, Sin 2.5, Pul 0.5, Pul 0.5 VF, Pul 1.5, and Pul 2.5, # representing significance with respect to the Static, Sin 0.5, Sin 1.5, Sin 2.5, x representing significance with respect to the Static, Sin 0.5, and Sin 1.5, + representing significance with respect to Static, Sin 0.5, Sin 1.5, Sin 2.5, and Pul 0.5 and open diamond representing significance with respect to the Static, Sin 0.5 and Sin 1.5. (b) F-actin fibres alignment induced by 24 h of WSS stimuli with * representing significance with respect to the Static and to Sin 0.5, and # representing significance with respect to the Static.
As expected, F-actin distribution was also affected by the different flow conditions (Fig. 3). Static controls show f-actin filaments distributed mostly at the cell periphery and randomly organized. EC exposed to Sin 0.5 show a distribution that is similar to the static control. However, these EC show what appear to be an immature formation of stress fibres within the cell body. By increasing the WSS peak in the presence of a reverse phase (Sin 1.5 and Sin 2.5) F-actin distribution becomes more structured but only partially organized. ECs exposed to pulsatile WSS profiles, show an organized cytoskeleton, with fully formed stress fibres crossing the cell body as previously described in the bumper-car mechanotransduction model.43
The quantified cell morphology show that EC exposed to pulsatile WSS waveforms are generally more elongated compared to exposure to sinusoidal WSS or maintained in static culture (Fig. 3a). The results were statistically significant for all the pulsatile conditions and the Sin 2.5 waveform if compared to the static control. We found that by doubling the TWSSG at the same peak magnitude, we induced a further significant elongation in all but Pul 0.5 VF, a waveform with a TWSSG that is four times higher than the original Pul 0.5, where EC have a reduced elongation compared to those exposed to Pul 0.5 or Pul 0.5 F.
The quantification of f-fibres direction shows an amplified aligned structure with the increase of WSS peak (Fig. 3b). Pulsatile conditions with a peak of 1.5 or 2.5 Pa induced a significant increase in alignment with respect to the static control and with respect to Sin 0.5. Pulsatile waveforms Pul 0.5 F, Pul 0.5 VF the Sin 2.5 profile results were only significant respect to the static controls. No differences were found with increasing TWSSG and the presence of a sinusoidal phase prevented the reorganization if compared to the waveforms having the same peak but lacking in the reverse phase.
We set out to examine the expression of genes coding for proteins involved in EC quiescence or activation and in the endothelial to mesenchymal transition (EndMT). Tested WSS profiles induced different regulations of several genes (Fig. 4) possibly involved in the flow-induced vascular lesion development.

KLF-2 and NFkB-3 (p65) immunofluorescence images of HUVECs after 24 h of exposure to the different WSS stimuli. Scale bar 20 μm. (a to d) Gene expression fold change vs. the static control presented in an arbitrary unit (a.u) for RT-PCR results and the protein expression vs. GAPDH expression control in a.u. (a) KLF-2 RT-PCR results with * representing significance with respect to Sin 0.5, Pul 0.5, Pul 1.5, Pul 0.5 F and Pul 0.5 VF. # representing significance with respect to Sin 0.5, Pul 0.5, Pul 0.5 F and Pul 0.5 VF, and Sin 2.5. open diamond representing significance with respect to Pul 2.5 (b) KLF-2 WB results with * representing significance with respect to the Static. # representing significance with respect to the Sin 0.5. x representing significance with respect to the Sin 2.5. + representing significance with respect to the Sin 1.5. (c) NFkB-3 RT-PCR results. (d) NFkB-3 WB results.
To evaluate EC activation in terms of activation or quiescence, we stained for KLF-2 and NFkB-3 (p65) to determine if different WSS profiles affect the expression and distribution of these proteins (Fig. 4).
ECs used as static control present a very weak KLF-2 signal located in the cell nuclei. ECs exposed Sin 0.5, show a slightly higher signal, not preferentially located in the nuclei. Post-exposure, ECs show the accumulation of KLF-2 protein in the nuclei, but still persistent in the cell body. Interestingly, all sinusoidal conditions induced a high dotted-shape signal nearby the cell nuclei, not present in the other conditions. ECs exposed to pulsatile waveforms have a positive signal preferentially located in the cell nuclei. Staining intensity was higher for profiles with a peak of 1.5 and 2.5 Pa compared to those stimulated at 0.5 Pa. Furthermore, Pul 0.5 VF-induced KLF-2 is very similar to the static. Consistent with the results of immunofluorescence analysis, KLF-2 gene expression up-regulated (Fig. 4a) after exposure to waveforms with a peak of 1.5 Pa or higher. Among 0.5 Pa profiles, only Pul 0.5 elicited an upregulation of KLF-2 gene. The increase in TWSSG caused a decrease in KLF-2 expression but not significant. The presence of a reverse phase did not reduce the gene expression if compared to the waveforms with the same positive peak. The quantification of KLF-2 protein expression (Fig. 4b) showed similar results if compared to the gene expression. We found that KLF-2 is upregulated by the waveforms with a peak of 1.5 Pa or higher. Pul 0.5 resulted the only 0.5 waveforms capable of inducing an upregulation of the protein even if to a lower level if compared to higher peak profiles. Interestingly, the presence of a reverse phase significantly increased the expression of this protein for WSS profiles with a peak of 1.5 Pa or higher.
As expected, flow-induced NF-kB expression induced by pulsatile profiles caused a nuclear translocation of the protein compared to the static controls (Fig. 4). Furthermore, as expected, Sin 0.5 did not induce the protein translocation and, surprisingly, neither did Sin 1.5 nor Sin 2.5. For protein quantification, we performed Western blotting. NFkB-3 gene expression (Fig. 4c) resulted similar after the exposure to all flow conditions and not significantly different if compared to the static controls. Similarly, NFkB-3 protein expression (Fig. 4d) resulted not affected by any flow conditions or by the static culture.
To evaluate further the activation state of EC exposed to different flow conditions, additional genes involved in the vascular tone control and in the pro-inflammatory pathways were quantified (Fig. 5). We found that nitric oxide synthase 3 (NOS-3 or e-NOS), a precursor of nitric oxide that is a potent vasodilation agent, gene expression was upregulated by all flow conditions. WSS peak, TWSSG and the presence of a reverse phase did not affect the expression. KLF-4 gene expression resulted upregulated by WSS waveforms with a peak of 1.5 Pa or higher and by Pul 0.5. No effects of varying TWSSG and reverse phase were found. Vascular adhesion molecule 1 (VCAM-1) and Interleukin-8 (Il-8) were downregulated by all conditions but Pul 0.5 VF. In particular, WSS profiles with a peak of 1.5 Pa or higher showed lower levels of these genes if compared to 0.5 Pa profiles. These results are significant if compared to Pul 0.5 VF. Both TWSSG and the reverse phase showed limited effects on the expression of the genes coding for these proteins.

Gene expression fold change vs. the static control presented in an arbitrary unit (a.u) for RT-PCR results. (a) KLF-4 RT-PCR results with * representing significance with respect to Sin 0.5, Pul 0.5, Pul 0.5 F and Pul 0.5 VF. # representing significance with respect to Sin 0.5, Pul 0.5 F, and Pul 0.5 VF. § representing significance with respect to Sin 0.5, Pul 0.5 F and Pul 0.5 VF. (b) eNOS-3 RT-PCR results. (c) vWF RT-PCR results. (d) VCAM-1 RT-PCR results with * representing significance with respect to Sin 2.5, Pul 1.5, Pul 1.5 F and Pul 2.5. (e) IL-8 RT-PCR results with * representing significance with respect to Sin 2.5, Pul 1.5, Pul 2.5 and Pul 2.5 F. (f) CD-144 (VE-Cadherin) expression.
Finally, markers of EndMT were quantified in terms of genes and proteins expression (Fig. 6). Cluster of differentiation 31 (CD-31 or PECAM), an endothelial identity marker, resulted significantly upregulated by Pul 1.5 F and Pul 2.5 F (Figs. 6a and 6b). The expression of this gene resulted in limited effects by different flow conditions compared to static controls. No differences were found for the expression of the cluster of differentiation 144 (CD144 or VE-Cadherin) and von Willebrand factor (vWF) (Figs. 5c and 5f), two endothelial identity markers. The α-SMA gene and protein expression, a marker of EndMT, was consistently upregulated by all flow conditions if compared to the static controls (Figs. 6c and 6d). However, no significantly differences were found among EC exposed to different WSS waveforms.

(a–d) Gene expression fold change vs. the static control presented in an arbitrary unit (a.u) for RT-PCR results and the protein expression vs. GAPDH expression control in a.u. (a) CD31 RT-PCR results with * representing significance of Pul 2.5F with respect to Pul 0.5 and Pul 1.5. # representing significance of Pul 1.5F with respect to Sin 0.5, Pul 0.5, Pul 0.5F and Pul 1.5. (b) CD-31 WB results. (c) α-SMA RT-PCR results. (d) α-SMA WB results.
Discussion
Endothelial cells control acute vascular tone and chronic vascular remodelling. These processes are regulated to maintain physiological mechanical stresses within the vascular system. EC possess different mechanisms to detect distinct features of flow profiles and regulate vessel physiology and remodelling accordingly.2
Numerous in-vivo and in-vitro evidences report OSI as a poor predictor of atherosclerotic lesion development in arteries.31,32 However, several computational studies performed in realistic AVF geometries still report OSI index in the analysis,14 therefore we considered this index to describe our WSS patterns.
Limited evidences are provided for the venous context since in-vitro experiment using HUVECs focused on arterial derived WSS.26 A lack of understanding of the role of specific hemodynamic parameters in the failure of AVF served as the motivation for this study.
After WSS exposure, morphological adaptations show that EC exposed to pulsatile waveforms elongate more than those maintained static or exposed to sinusoidal WSS. By increasing the WSS peak, it is possible to induce elongation further. Increasing the TWSSG also induces a significant increase in elongation. However, this seems not to depend on a specific gradient. F-actin fibres alignment depend on WSS peak more than on TWSSG. Alignment achieved by ECs exposed to WSS waveforms with a peak of 1.5 Pa or higher significantly increased in compared to Pul 0.5 profile. The presence of a reverse phase reduced the levels of alignment compared to pulsatile counterparts. However, F-actin fibres alignment showed an increasing alignment trend with increasing WSS peak, being significant for Sin 2.5 despite the presence of the reverse phase.
In terms of EC phenotype, the peak of the WSS waveforms is the main feature in inducing a protective effect in ECs in culture. KLF-2 ad KLF-4 are two known flow-induced proteins involved in ECs quiescence, cytoskeleton remodelling and are therefore considered7 protective. Both KLF-2 and KLF-4 were up-regulated by waveforms with a peak of 1.5 Pa or higher. This up-regulation was consistently independent of the reverse phase (OSI value) or the TWSSG. It is important to note that high levels of gene and protein expression were found after exposure to waveforms averaging less than 0.5 Pa, a low value previously related to the atheroprone phenotype in EC. It is also worth noting that in terms of localisation, KLF-2 shows a unique distribution in ECs stimulated with sinusoidal WSS, while after pulsatile shear and in static control (with a lower intensity), KLF2 appears to be strictly located at the nuclear level. EC exposed to sinusoidal waveforms have a dotted-shape localization adjacent to the nuclei. Very few studies have described endothelial cell KLF-2 immunofluorescence in vitro17 and further studies are required to confirm the phenomenon and to relate this localisation to a specific biological significance.
Regarding pro-inflammatory gene expression, we found no differences in the expression of the p-65 subunit of NFkB (NFkB-3). The localisation of this protein however is strongly affected by the nature of WSS waveforms and it is in agreement with previous findings15 that show how protective WSS induces the translocation of the protein from the nucleus preventing the activation of pro-inflammatory pathways. Sinusoidal WSS, on the contrary, did not affect NF-kB localisation with respect to the static control. While this was expected for Sin 0.5, it was not for Sin 1.5 and especially Sin 2.5. This is also in contrast with the levels of KLF-2 that show high expression in both Sin 1.5 and Sin 2.5. This distribution may be related to the shortened time of WSS exposure or an additional biological effect, and thus remains to be elucidated.
In general, the protective effect of high-peak WSS patterns is further confirmed by the downregulation of adhesion molecule VCAM-1 and pro-inflammatory signal IL-8.27,38 On the contrary, these genes resulted up-regulated by waveforms with a peak of 0.5 Pa. In this case, the increase of TWSSG caused an increased expression, significantly stronger for Pul 0.5 VF.
WSS waveforms with a peak of 0.5 Pa showed to have peculiar effects. Pul 0.5 profile induced a relative protective phenotype by inducing a limited up-regulation of KLF-2 and KLF-4. This can be reversed by the presence of a 0.5 Pa reverse phase and furthermore by increase of the TWSSG. Despite TWSSG only partially affected cell elongation and alignment, exposure to Pul 0.5 F and especially Pul 0.5 VF caused a strong KLF-2 downregulation and an increased expression of pro-inflammatory signals such as IL-8 and VCAM-1. It is worth noticing that Sin 0.5 and Pul 0.5 VF present similar low protective activity in terms of KLF-2, KLF-4 and NF-kB expression. However, Pul 0.5 VF showed higher expression for IL-8 and VCAM-1 resulting more susceptible even at higher TAWSS and lower OSI in comparison to Sin 0.5. This is probably due to the lack or the insufficient stimuli for EC to establish a clear preferential direction. However, a comprehensive explanation for these results remain unfulfilled.
EndMT causes ECs to switch phenotype to a more proliferative and secretory state becoming myofibroblasts.13,48 Subsequently, myofibroblasts are the main cellular component of neo-intimal hyperplasia even though it remains unknown which vascular cells are responsible for the dysregulated growth of the vascular wall. The retention of the endothelial identity by EC exposed to different flow conditions is shown by the stable expression of gene coding for CD-31, CD-144 and vWF, three markers of the endothelial phenotype39 that are not affected by the experimental conditions. This aspect however is in contrast with the up-regulation of the α-SMA, a marker for EndMT that is observed for gene and protein expression, even if not statistically significant.. It is worth noting that all the experiments were conducted in the absence of exogenous TGF-β that is usually used to study EndMT in-vitro.
A WSS feature that is only partially addressed in this work but that can be important in understanding the role of hemodynamics in vascular pathologies is the frequency content of WSS profiles. Feaver and colleagues showed that is possible to reverse the protective effect of unidirectional WSS patterns by increasing the magnitude of higher frequency component.21 The increase in TWSSG is only a component of the increase of frequency content. We showed that increasing TWSSG without affecting the overall pattern frequency does not have significant effects on ECs. Further research activities need to address systematically the WSS frequency content effects on EC.
In conclusion, when assessing the effects of unsteady WSS profiles, the previously considered atherogenic TAWSS threshold should be reconsidered and reduced significantly, at least for venous EC. This study focused on the response of venous EC to specific hemodynamic parameters of AVF. The aetiology of atherosclerosis and other flow-induced arterial pathologies is very different from the causes of stenosis in venous tissues connected to arterial vasculature. Direct relation between flow induced venous remodelling and arterial lesions development requires careful consideration. We demonstrated that WSS profiles averaging 0.28 Pa (Pul 1.5 F) is capable of inducing a strong KLF-2 and KLF-4 up regulation. Similarly, waveforms with an average of 0.16 Pa (Pul 0.5) induce the upregulation of protective genes even if to a lower extent. Only Sin 0.5 and especially Pul 0.5VF failed to induce the protective EC phenotypes. These results indicate that both reverse phase and time-averaged WSS might yet provide information about the risk associated with specific WSS waveforms but both indices must be considered in conjunction with additional components of the stimuli and especially with the WSS peak. Considering clinical aspects, arteriovenous fistula’s venous tissues that do not display an increase in diameters within 24 h predict the failure of the vascular access in almost 60% of the cases.11
By informing computational models with accurate prediction of the downstream effects of hemodynamics on vascular biology, it will be possible to assess and predict the outcome of vascular pathologies in a non-invasive and precise fashion.
References
Ajami, N. E., S. Gupta, M. R. Maurya, P. Nguyen, J. Y.-S. Li, J. Y.-J. Shyy, Z. Chen, S. Chien, and S. Subramaniam. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc. Natl. Acad. Sci. USA 114:10990–10995, 2017.
Baeyens, N., C. Bandyopadhyay, B. G. Coon, S. Yun, and M. A. Schwartz. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Invest. 126:821–828, 2016.
Boon, R. A., T. A. Leyen, R. D. Fontijn, J. O. Fledderus, J. M. C. Baggen, O. L. Volger, G. P. van Nieuw Amerongen, and A. J. G. Horrevoets. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 115:2533–2542, 2010.
Brahmbhatt, A., A. Remuzzi, M. Franzoni, and S. Misra. The molecular mechanisms of hemodialysis vascular access failure. Kidney Int. 89:303–316, 2016.
Browne, L. D., K. Bashar, P. Griffin, E. G. Kavanagh, S. R. Walsh, and M. T. Walsh. The role of shear stress in arteriovenous fistula maturation and failure: a systematic review. PLoS ONE 10:e0145795, 2015.
Bryan, M. T., H. Duckles, S. Feng, S. T. Hsiao, H. R. Kim, J. Serbanovic-Canic, and P. C. Evans. Mechanoresponsive networks controlling vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 34:2199–2205, 2014.
Chang, E., L. Nayak, and M. K. Jain. Krüppel-like factors in endothelial cell biology. Curr. Opin. Hematol. 24:224–229, 2017.
Chatterjee, S. Endothelial mechanotransduction, redox signaling and the regulation of vascular inflammatory pathways. Front. Physiol. 9:524, 2018.
Chatzizisis, Y. S., A. U. Coskun, M. Jonas, E. R. Edelman, C. L. Feldman, and P. H. Stone. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 49:2379–2393, 2007.
Cheng, M., J. Wu, X. Liu, Y. Li, Y. Nie, L. Li, and H. Chen. Low shear stress-induced interleukin-8 mRNA expression in endothelial cells is mechanotransduced by integrins and the cytoskeleton. Endothelium 14:265–273, 2007.
Cheung, A. K., P. B. Imrey, C. E. Alpers, M. L. Robbin, M. Radeva, B. Larive, Y.-T. Shiu, M. Allon, L. M. Dember, T. Greene, J. Himmelfarb, P. Roy-Chaudhury, C. M. Terry, M. A. Vazquez, J. W. Kusek, H. I. Feldman, and Hemodialysis Fistula Maturation Study Group. Intimal hyperplasia, stenosis, and arteriovenous fistula maturation failure in the hemodialysis fistula maturation study. J. Am. Soc. Nephrol. 28:3005–3013, 2017.
Chistiakov, D. A., A. N. Orekhov, and Y. V. Bobryshev. Effects of shear stress on endothelial cells: go with the flow. Acta Physiol. 219:382–408, 2017.
Cho, J. G., A. Lee, W. Chang, M.-S. Lee, and J. Kim. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 9:294, 2018.
Cunnane, C. V., E. M. Cunnane, and M. T. Walsh. A review of the hemodynamic factors believed to contribute to vascular access dysfunction. Cardiovasc. Eng. Technol. 8:280–294, 2017.
Dai, G., M. R. Kaazempur-Mofrad, S. Natarajan, Y. Zhang, S. Vaughn, B. R. Blackman, R. D. Kamm, G. García-Cardeña, and M. A. Gimbrone. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. USA 101:14871–14876, 2004.
Davies, P. F., M. Civelek, Y. Fang, and I. Fleming. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 99:315–327, 2013.
Dekker, R. J., R. A. Boon, M. G. Rondaij, A. Kragt, O. L. Volger, Y. W. Elderkamp, J. C. M. Meijers, J. Voorberg, H. Pannekoek, and A. J. G. Horrevoets. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107:4354–4363, 2006.
Dowling, C. M., D. Walsh, J. C. Coffey, and P. A. Kiely. The importance of selecting the appropriate reference genes for quantitative real time PCR as illustrated using colon cancer cells and tissue. F1000Research 5:99, 2016.
Ene-Iordache, B., L. Mosconi, L. Antiga, S. Bruno, A. Anghileri, G. Remuzzi, and A. Remuzzi. Radial artery remodeling in response to shear stress increase within arteriovenous fistula for hemodialysis access. Endothelium 10:95–102, 2003.
Ene-Iordache, B., and A. Remuzzi. Disturbed flow in radial-cephalic arteriovenous fistulae for haemodialysis: low and oscillating shear stress locates the sites of stenosis. Nephrol. Dial. Transplant. 27:358–368, 2012.
Feaver, R. E., B. D. Gelfand, and B. R. Blackman. Human haemodynamic frequency harmonics regulate the inflammatory phenotype of vascular endothelial cells. Nat. Commun. 4:1525, 2013.
Fledderus, J. O., R. A. Boon, O. L. Volger, H. Hurttila, S. Ylä-Herttuala, H. Pannekoek, A.-L. Levonen, and A. J. G. Horrevoets. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28:1339–1346, 2008.
Fledderus, J. O., J. V. van Thienen, R. A. Boon, R. J. Dekker, J. Rohlena, O. L. Volger, A.-P. J. J. Bijnens, M. J. A. P. Daemen, J. Kuiper, T. J. C. van Berkel, H. Pannekoek, and A. J. G. Horrevoets. Prolonged shear stress and KLF2 suppress constitutive proinflammatory transcription through inhibition of ATF2. Blood 109:4249–4257, 2007.
Franzoni, M., I. Cattaneo, B. Ene-Iordache, A. Oldani, P. Righettini, and A. Remuzzi. Design of a cone-and-plate device for controlled realistic shear stress stimulation on endothelial cell monolayers. Cytotechnology 68:1885–1896, 2016.
Franzoni, M., I. Cattaneo, L. Longaretti, M. Figliuzzi, B. Ene-Iordache, and A. Remuzzi. Endothelial cell activation by hemodynamic shear stress derived from arteriovenous fistula for hemodialysis access. Am. J. Physiol. Heart Circ. Physiol. 310:49–59, 2015.
Franzoni, M., and M. T. Walsh. Towards the identification of hemodynamic parameters involved in arteriovenous fistula maturation and failure: a review. Cardiovasc. Eng. Technol. 8:342–356, 2017.
Koh, Y., and J. Park. Cell adhesion molecules and exercise. J. Inflamm. Res. 11:297–306, 2018.
Liao, J. K. Hindsight Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 123:540–541, 2013.
Lu, D. Y., E. Y. Chen, D. J. Wong, K. Yamamoto, C. D. Protack, W. T. Williams, R. Assi, M. R. Hall, N. Sadaghianloo, and A. Dardik. Vein graft adaptation and fistula maturation in the arterial environment. J. Surg. Res. 188:162–173, 2014.
Malek, A. M., S. L. Alper, and S. Izumo. Hemodynamic shear stress and its role in atherosclerosis. JAMA 282:2035, 1999.
Mohamied, Y., E. M. Rowland, E. L. Bailey, S. J. Sherwin, M. A. Schwartz, and P. D. Weinberg. Change of direction in the biomechanics of atherosclerosis. Ann. Biomed. Eng. 43:16–25, 2015.
Mohamied, Y., S. J. Sherwin, and P. D. Weinberg. Understanding the fluid mechanics behind transverse wall shear stress. J. Biomech. 50:102–109, 2017.
Moore, Jr, J. E., C. Xu, S. Glagov, C. K. Zarins, and D. N. Ku. Fluid wall shear stress measurements in a model of the human abdominal aorta: oscillatory behavior and relationship to atherosclerosis. Atherosclerosis 110:225–240, 1994.
Morton, S. K., A. J. Rodríguez, D. R. Morris, A. P. Bhandari, J. V. Moxon, and J. Golledge. A systematic review and meta-analysis of circulating biomarkers associated with failure of arteriovenous fistulae for haemodialysis. PLoS ONE 11:0159963, 2016.
Nakajima, H., and N. Mochizuki. Flow pattern-dependent endothelial cell responses through transcriptional regulation. Cell Cycle 16:1893–1901, 2017.
Parmar, K. M., H. B. Larman, G. Dai, Y. Zhang, E. T. Wang, S. N. Moorthy, J. R. Kratz, Z. Lin, M. K. Jain, M. A. Gimbrone, and G. García-Cardeña. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Invest. 116:49–58, 2006.
Peiffer, V., S. J. Sherwin, and P. D. Weinberg. Does low and oscillatory wall shear stress correlate spatially with early atherosclerosis? A systematic review. Cardiovasc. Res. 99:242–250, 2013.
Poredos, P., and M. K. Jezovnik. The role of inflammation in venous thromboembolism and the link between arterial and venous thrombosis. Int. Angiol. 26:306–311, 2007.
Rakocevic, J., D. Orlic, O. Mitrovic-Ajtic, M. Tomasevic, M. Dobric, N. Zlatic, D. Milasinovic, G. Stankovic, M. Ostojić, and M. Labudovic-Borovic. Endothelial cell markers from clinician’s perspective. Exp. Mol. Pathol. 102:303–313, 2017.
Remuzzi, A., M. Bozzetto, and P. Brambilla. Is shear stress the key factor for AVF maturation? J. Vasc. Access 18:S10–S14, 2017.
Remuzzi, A., B. Ene-Iordache, L. Mosconi, S. Bruno, A. Anghileri, L. Antiga, and G. Remuzzi. Radial artery wall shear stress evaluation in patients with arteriovenous fistula for hemodialysis access. Biorheology 40:423–430, 2003.
Tabas, I., G. García-Cardeña, and G. K. Owens. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 209:13–22, 2015.
Thi, M. M., J. M. Tarbell, S. Weinbaum, and D. C. Spray. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a bumper-car model. Proc. Natl. Acad. Sci. USA 101:16483–16488, 2004.
Tzima, E. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ. Res. 98:176–185, 2006.
Wentzel, J. J., Y. S. Chatzizisis, F. J. H. Gijsen, G. D. Giannoglou, C. L. Feldman, and P. H. Stone. Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc. Res. 96:234–243, 2012.
Wójciak-Stothard, B., S. Potempa, T. Eichholtz, and A. J. Ridley. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 114:1343–1355, 2001.
Wojciak-Stothard, B., and A. J. Ridley. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J. Cell Biol. 161:429–439, 2003.
Xiao, L., and A. C. Dudley. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 241:25–35, 2017.
Acknowledgments
The authors would like to thank Science foundation Ireland (SFI) (Grant No. 15/CDA/3323) and the Mid-Western Cancer Foundation for providing financial support to this project.
Conflict of interest
The authors declare no conflicts of interest, financial or otherwise.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Umberto Morbiducci oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Franzoni, M., O’Connor, D.T., Marcar, L. et al. The Presence of a High Peak Feature Within Low-Average Shear Stimuli Induces Quiescence in Venous Endothelial Cells. Ann Biomed Eng 48, 582–594 (2020). https://doi.org/10.1007/s10439-019-02371-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-019-02371-5