
Abstract
Mechanical stress has been proven to be an important factor interfering with many biological functions through mechano-sensitive elements within the cells. Despite the current interest in mechano-transduction, the development of suitable experimental tools is still characterized by the strife to design a compact device that allows high-magnification real-time imaging of the stretched cells, thus enabling to follow the dynamics of cellular response to mechanical stimulations. Here we present a microfluidic multi-layered chip that allows mechanical deformation of adherent cells maintaining a fixed focal plane, while allowing independent control of the soluble microenvironment. The device was optimized with the aid of FEM simulation and fully characterized in terms of mechanical deformation. Different cell lines were exposed to tunable mechanical strain, which results in continuous area deformation up to 20%. Thanks to the coupling of chemical glass etching, 2-dimensional deformation of a thin elastomeric membrane and microfluidic cell culture, the developed device allows a unique combination of cell mechanical stimulation, in line imaging and accurate control of cell culture microenvironment.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recent discoveries have proven the role of mechanical stress in cellular behaviour and fate. More specifically external mechanical stimuli have an enormous impact in the correct development and functionality of tissues and organs6,13,18,19,20,25 influencing, among others, crucial developmental steps such as actin fibers organization1 and the endothelial-mesenchymal transition through the activation of different pathways accordingly to strain magnitude.6 Abnormal mechanical strain is correlated also with the pathogenesis of specific tissues, such as cardiac and muscle tissue.21,28,31 Moreover, it plays a relevant role in the activation of mechano-sensitive pathways such as the Hippo signalling pathway5 and the local angiotensin signalling system.15
In this context the fast-growing field of mechano-transduction arises, describing the cellular processes that translate mechanical stimuli into biochemical signals, thus enabling cells to adapt to their physical surroundings.
Stretch-induced responses appear not only at hours or days from the beginning of the stimulus, but also at shorter time scales such as seconds to minutes.8 The earliest events in response to mechanical stress, which generally occur at the cell surface in the proximity of sites of mechano-sensation, can include opening of stretch-activated cation channels,24 ions fluxes, induction of enzymatic activity of integrin-associated signalling molecules,27 induction of heterotrimeric G-proteins14 and production of reactive oxygen species.23
The investigation of such mechanisms occurring during or immediately after an imposed static or dynamic mechanical stretch requires a real time imaging of the cells, that often is not taken into account for long-term experiments. As an example, Kolsmaska et al. showed that in response to stretch, before any active response, cell membranes adapt almost instantaneously (seconds to minutes) through a passive mechanism generating invaginations to continuously store and release large fractions of the cell membrane, minimizing its elastic and adhesion energies.17 Calcium transient occurs also very quickly, with a time scale of few seconds. In beating cardiomyocytes a physiologic stretch of 8% rapidly triggers a burst of Calcium ions sparks, regulated by a local production of reactive oxygen species, which is impaired in pathologic conditions.23 Similarly, epithelial wound healing triggers in a very short time a series of cell responses, including calcium fluxes26 and rapid cytoskeleton remodelling.4
Currently, several devices to stretch cultured cells are available—both commercial and custom made.12,16 The commercial devices most commonly used to culture cells in mechanically active conditions are the ones produced by Flexcell International Corporation that relay on a elastomeric membrane fixed on a O-ring in which vacuum is obtained by stretching the membrane.18,25,29 However the main drawback of such a device is its size. Another common method used to stretch cultured cells employs an external servomotor that physically pulls the elastomeric membrane (along one or two directions) to obtain deformation.1,6,7,22 Most of them relay on external systems to exert the mechanical stimulation such as servo-motors and do not allow real time imaging of the stretched cells.
Other possible methods to simulate stretching conditions involve the use of biochemical signals reproducing those secerned by cells during mechanical strain,10 or the exploitation of osmotic stress. Compared to these solutions, the use of pneumatic actuation has the advantage to induce homogeneous mechanical strain without interfering with cells, even though coupling cell stretching with on line analysis can be extremely difficult.
In addition, miniaturized systems and microfluidics can greatly enhance the control of cellular microenvironment. For example, it would allow the coupling of stimuli such as shear stress with deformation to reproduce in vitro phenomena experienced by endothelial cells in a blood vessel in both physiologic and pathologic conditions.3,9
However, stretching devices completely based on the microfluidic technology and easy coupling with microscopes for on line analysis have been not completely exploited yet.12 Recently, we have developed a microfluidic stretching platform for investigating the role of Hippo signalling pathway in mechano-transduction under active mechanical stimulation5 that employed a microfluidic inflatable membrane. The technology developed could be further improved for studying the dynamic of biological response under cyclic mechanical stimulation.
In this work, we present a microfluidic platform that allows accurate mechanical stimulation of cultured cells while maintaining the focal plane, taking up a minimal amount of space. Both the culture chamber and the stretching system are integrated in the same multi-layered microfluidic device allowing for accurately controlled experimental conditions of both biochemical and mechanical stimuli and fast analysis of mechano-transduction-associated cell responses.
Two different prototypes were realized varying the width of the stretchable area, which also affect the maximum achievable deformation. The innovative stretching mechanism we propose allows stimulating cultured cells while maintaining a fixed focal plane and can be coupled with any microfluidic chamber and optical system, including a confocal microscope. Moreover, we tested the system using a Actin-EGFP reporter cell line and imaged actin filaments network in real time during cell mechanical deformation.
This design will enable to observe in real-time and at high resolution the dynamics of cellular response to external mechanical cues; at the same time the cellular soluble environment can be accurately controlled, while enlarging the experimental variables affecting mechano-transduction response to be tested. This allows to study rapid events in response to mechanical stretch, in particular those related to conformational changes of cell components, such as the membrane or cytoskeletal tension.
Materials and Methods
Glass Wet Etching
Standard microscopy slides (75 × 25 mm) made in borosilicate glass were washed with Micro90 cleaning solution (Sigma-Aldrich), rinsed with deionized water and dried using a nitrogen flux. Positive photoresist (SPR227, ROHM and HAAS) was deposited on the glass surface following the manufacturer instructions and the serpentine-shaped system of channels was impressed on the photoresist through UV exposure (AOI lamp) using a photo-mask. After photoresist development the slides were left resting overnight to promote resist adhesion, then the rear of the glass slide was protected with tape to proceed with the etching process.
The glass slides were dipped in a Petri dish containing hydrofluoric acid at 40% weight-based concentration (Sigma-Aldrich) for 3 min, and put on an oscilloscope (Yellowline, IKA) to promote mass transfer of acid to the reacting glass surface. Glass slides were washed once with deionized water, once with sulphuric acid at 98% weight-based concentration (Sigma) and again once with deionized water. Acetone (Sigma-Aldrich) was used in order to remove the photoresist and the tape. Finally the microscopy slides were cleaned using a piranha solution, made by one part hydrogen peroxide at 30% concentration (Sigma-Aldrich) and three parts of sulphuric acid at 98% concentration (Sigma-Aldrich), to remove residuals from etching reaction.
Glass Thermal Treatment
Slides were heated at fixed temperature (690, 700, 710 or 720 °C) for 30 min with a heating rate of 5 °C/min. Cooling occurred naturally in order to avoid the presence of residual thermal stresses in the glass.
Glass Surface Analysis
Microscopy slide surface was analysed using an optical 3D profiler (PluNeox, Sensofar) set in confocal mode at × 20 and × 100 magnification to assess the surface roughness, by means of parameters Ra and Rz, and inspect the channels cross section after thermal treatment carried out at different temperatures. Preliminary measurements showed that the profile was homogeneous along the channels so it was sufficient to acquire and analyse just a portion of the channel. Due to the high magnification, portions of the serpentine cross section were individually acquired and eventually stitched together to obtain the complete picture.
Elastic Membrane Production
The elastomeric membrane was fabricated in poly(dimethylsiloxane), PDMS, (Sylgard 184, Down Corning) using a 1:10 weighted ratio of curing agent and pre-polymer. The liquid PDMS was extensively degassed in a vacuum container to remove all air bubbles and then poured on a 125 mm diameter silicon wafer previously treated with hexamethyldisiloxane, HMDS, (Sigma-Aldrich) to promote the following PDMS detachment. After a thermal preconditioning at 37 °C on a levelled hotplate (Falc) the wafer was put on a spin coater (Laurell) and spun for 5 s at 500 rpm final speed and 100 rpm/s acceleration and then for 1 min at 1400 rpm with acceleration of 100 rpm/s in order to achieve a membrane thickness of 40 µm. The membrane was finally baked on a levelled hot plate (Falc) at 80 °C for 50 min and removed from the wafer.
Microfluidic Layer Fabrication
The microfluidic mould was fabricated using standard soft-lithography techniques. The channel geometry was impressed on a 100 mm diameter silicon wafer using a negative photoresist (SU8-2050, Microchem) and developing it after UV exposure (AOI lamp) with a custom-made photo-mask. All procedures were conducted accordingly to the producer user’s manual, in order to achieve a final height of 200 µm.
The microfluidic mould was realized in PDMS (Sylgard 184, Down Corning) with a 1:10 weighted ratio of curing agent and pre-polymer. The liquid PDMS was poured on the wafer, degassed in a vacuum chamber and cured in a preheated oven at 70 °C for 2 h. After the crosslinking reaction was completed the PDMS mould was extract from the wafer.
Device Assembly and Functionalization
The three different layers were produced separately and then assembled in the final device. Firstly the 40 µm-thick membrane was attached to the microfluidic mould via surface plasma activation (Harrick Plasma). Subsequently the PDMS layers were attached to the glass slides using the plasma bounding technique as well. In this latter case it was necessary to bind only the periphery while the centre of the device (comprising the central flat area devoted to cell culturing and the etched serpentines) had to be left free to slide. In order to selectively bind the layers, only the membrane external surface was exposed to the plasma treatment.
Once the device was assembled, Tygon tubings (Cole-Parmer) were connected to the serpentine circuit and it was filled with PBS solution (Thermo Fisher Scientific). In order to remove air bubbles the circuit was pressurized at 0.5 bar.
The device was then functionalized for cell culture. Firstly the bottom of the device was sterilized using 70% weight based concentration ethanol (Carlo Erba), then surface was sterilized via UV exposure for 30 min and finally of the microfluidic chamber was sterilized flushing ethanol and subsequently washed with sterile deionized water and left drying.
After complete drying the microfluidic chamber was incubated at 37 °C, 95% humidity and 5% CO2 for 30 min with 50 µg/mL fibronectin solution (Sigma-Aldrich) to promote cells adhesion. The microfluidic chamber was rinsed flushing sterile PBS and cells were seeded with concentration of 1000 cell/µL.
Immediately before performing the stretching experiments the tubes were connected to two identical 250 µL glass syringes (Hamilton) and the syringes were placed in a coupled syringe pump (Texas Instruments) set in withdrawal mode with suctioning flow-rate set at 600 µL/min.
Numerical Simulation of Membrane Deformation
Numerical analyses of the glass-membrane mechanical interaction were developed by using a Finite Element software (ABAQUS®, SIMULIA, Daussault Systems), in order to obtain a suitable stretch of the PDMS membrane. Several aspects affecting PDMS membrane deformation were considered, such as the thickness of the membrane, the number, depth and width of the glass channels. Three values of membrane thickness were evaluated: 20, 30 and 40 μm. For each thickness, we considered a channel depth of 60 and 80 mm and a channel width of 160, 220 and 250 μm. For a limited set of geometries, it was considered the effect of the number of channels, varying this from 1 to 5 per side. A brief description of the combination of parameters considered is reported in Table 1. Analyses were developed both considering the nominal geometry of the glass and the effective geometry resulting from the etching. According to the geometry of glass and PDMS membrane, it was possible to investigate the mechanical problem by adopting two-dimensional models, assuming plane strain conditions and modelling only half of the structures. The contact between elastomeric membrane and glass was considered frictionless. The mechanical response of the PDMS was described by assuming an almost-incompressible neo-Hookean hyperelastic constitutive model. The constitutive model was fitted on the basis of tensile experimental tests developed on PDMS samples (Planar Biaxial TestBench Instrument, Bose® Electro-Force), with constitutive parameters corresponding to an initial longitudinal elastic modulus of 1.0 MPa.
Cell Culture
Human foreskin fibroblasts were expanded in 75-mL tissue culture flasks with high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM 41965, Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific). In order to verify cell viability, cells were incubated for 30 min with 3 µM calcein (Sigma-Aldrich) and analysed with a fluorescent microscope (Leica).
Human immortalized mammary epithelial cells MCF10A, stably expressing membrane-bound EGFP (Mb-EGFP) and nuclear-localized mCherry (NLS-Cherry), were kindly provided by Stefano Piccolo’s Lab (Department of Molecular Medicine, University of Padova, Italy) and cultured as described in Aragona et al.5 U2OS cells were purchased from Sigma-Aldrich (CLL1032) and cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM 41965, Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific). At 90% confluence, cells were trypsinized with 0.25% trypsin–EDTA (Thermo Fisher Scientific), centrifuged at 1200 rpm for 5 min, and re-suspended in growth medium for injection into the microfluidic device.
Results
Design of the Stretching Device
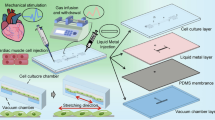
The operating principle of the cell stretching device is based on the elongation of a thin elastic membrane along a horizontal plane, which allows the maintenance of the focal plane of cells cultured above. This is obtained by pulling the membrane inside glass-etched micro-channels nearby the cell stretching area (Fig. 1a). An additional microfluidic layer containing a microfluidic chamber for cell culture was employed to obtain a confined environment in which medium composition can be accurately controlled both spatially and temporally.

(a) Working principle of the cell stretching device. (b) Stretching device is composed by three different layers: the glass slide with micro-channels derived through chemical etching; the elastic membrane where cells adhere and are exposed to deformation; an additional microfluidic layer with cell culture chamber. (c) Geometric details of the pneumatic circuit etched in the glass slide (left) and cell culture chamber (right). (d) Picture of the assembled cell stretching device.
The entire device is composed of three different layers, as schematically represented in Fig. 1b. The bottom layer consists in a microscopy glass slide containing two symmetric micro-channels network realized through glass chemical etching; the middle layer is composed by a thin elastomeric membrane on which cells are cultured in adhesion; the top layer is made by a microfluidic layer containing a micrometric chamber for cell culture. Mechanical deformation is achieved by generating a pressure difference (ΔP) between the two faces of the membrane, thus pulling the membrane inside the channels realized in the glass. This causes an active stretching, mainly in the direction orthogonal to the channels length, of cells cultured on the flat surface between the two channels network (Fig. 1b). The serpentine-shaped system of micro-channels etched in the glass is designed in order to enhance the maximum achievable stretching, as to multiply the deformation allowed by a single channel.
Figure 1c shows the geometric details of the stretching device prototype with corresponding dimensions. The whole microfluidic device (Fig. 1d) can be easily integrated in standard cell culture plates or coupled with a wide variety of microscopes, including a confocal microscope, for on line analysis of cell stretching and cell-deformation responses.
Numerical Simulation of Membrane Deformation
We implemented numerical simulations to investigate the effect of the design variables, namely membrane thickness, glass channel geometry and number of channels on the overall device performance. Table 1 summarizes the range of explored values by means of Finite Element modelling described in “Numerical Simulation of Membrane Deformation” section.
Figure 2a–2c shows the influence of channel shape and membrane thickness on membrane deformation. In particular, Fig. 2a shows the deformation of a 40 μm-thick membrane, placed above channels of different geometry and subjected to the same pressure difference of 1 bar. Wider channels with smooth edges allow higher membrane deformability and sliding.

(a) Vertical displacement (in μm) of a 40 μm-thick PDMS membrane on different shapes of the glass, subjected to 1 bar negative pressure applied to the lower surface. (b) Deformation of PDMS membranes with different thicknesses (20, 30 and 40 μm) on the same glass substrate, at negative pressure of 1 bar. The contours represent the maximum tensile logarithmic strain in percentage. (c) Maximum tensile logarithmic strain in the flat central region of the membranes as a function of the applied pressure using different thicknesses (20, 30 and 40 μm). (d) Deformation of a 40 μm-thick PDMS membrane placed on a glass substrate with an increasing number of channels per side and a flat central region of 250 μm width.
The effect of the membrane thickness on the deformation within the same channel is shown in Fig. 2b, with the same pressure difference of 1 bar. Reducing membrane thickness enhances its deformability and, consequently, the strain cells are subjected. As an example, the strain can be increased from 10 to 16% by halving the thickness from 40 to 20 µm. The graph of Fig. 2c shows the level of maximum strain obtained in the flat central region of the membrane versus the pressure applied for the three thicknesses used in the simulation of Fig. 2b.
The analysis indicates that membrane thickness and channel geometry contribute to define the achievable cell deformation. We verified that the actual glass profile obtained during the etching process is wide enough to allow for good deformability also of 40 µm-thick membranes. Therefore we decided to employ a 40 µm-thick membrane exploiting its greater strength to limit the possibility of membrane rupturing during assembly and deformation.
We also evaluated the influence of the number of channels in the serpentine in increasing the membrane deformation. Figure 2d shows the deformation profile of a 40 μm-thick membrane on the actual profile of a glass substrate with 5 channels per side and a flat 250 μm wide central surface. This simulation was intended to evaluate the maximum strain in the flat region of the membrane obtained by progressively applying the negative pressure from the external to the internal channels of the glass.
The analysis of the number of channels-dependent deformation reported in Fig. 2d validates the idea to multiply the channels to obtain greater values of deformation. For this we decided to realize 5 interconnected channels on each side of the central flat area.
Glass Surface Analysis and Thermal Treatment
The final set up of the device has channels with 80 µm depth and 220 µm width. However, as a consequence of chemical etching, the glass surface results very rough and sharp edges can be found on the etched channels walls, that may prevent membrane sliding during deformation or, even, damage of the membrane itself. In order to promote membrane sliding on the glass surface and avoid membrane damaging, we performed a thermal treatment on the etched microscopy slides to smooth glass surface.
Figure 3a shows that the temperature greatly affects the final outcome. The curvature of the channel measured at the bottom of the channel wall increases by increasing the temperature (Fig. 3b). Moreover, the higher the temperature the smoother result the channels, as revealed by decreased roughness indexes Ra and Rz (Fig. 3c). However, if the process temperature is too close to the glass softening temperature, channels shape is irremediably compromised. We identified an optimal temperature within the range of 700 and 710 °C, therefore a temperature of 700 °C was selected.

(a) Surface topography analysis using an optical 3D profiler after thermal treatment of the glass-etched channels at increasing temperatures. At T = 720 °C channels are almost completely collapsed. (b) Curvature angle measured at the bottom of the etched channel increases by increasing the softening temperature. (c) Surface roughness parameters (Ra, Rz) decreases by increasing the softening temperature, thus reducing friction on the membrane during deformation and the chances of rupture. Bars are expressed as mean ± SE.
This analysis shows that the thermal treatment greatly increases surface smoothness and therefore it is of crucial importance in the device manufacturing process.
Fluidic-Based Control of Membrane Deformation
The membrane deformation is achieved by generating a force that pulls the membrane inside the etched channels. This can be obtained by withdrawing the fluid from the serpentine-shaped channels using a syringe pump, thereby reducing the pressure on glass side.
In order to actually enhance maximum achievable deformation using a multiple channel structure, the channels must be emptied starting from the furthest from the centre and orderly go towards the central channels or at least simultaneously. In this way the membrane is gradually pulled, and thus deformed, as the channels are emptied.
In order to verify the correct fluid withdrawal, we filled the channels with 1 mM fluorescein solution and placed the device under a fluorescence microscope at × 5 magnification. The fluid was suctioned using a syringe pump equipped with two identical glass syringes of 250 µL volume set in withdrawal mode with a flow rate of 1 mL/min. As can be seen in Fig. 4, in rest position (t = 0 s) the channels are completely filled with fluorescent dye. As the withdrawal begins, the channels are emptied simultaneously until only residual fluorescence intensity can be observed (Fig. 4).

Validation of the pneumatic circuit. The circuit was filled with a fluorescent dye and then emptied, verifying that the fluorescence intensity decreases homogeneously among channels until only a low fluorescent signal is detected.
These results showed the correct and complete emptying of the serpentine circuit confirming that the multiple channels structure can actually enhance total deformation achievable.
Cell Stretching
In order to perform cell stretching experiments, we first tested the cell viability within the device after 2 weeks of culture using human fibroblasts. After staining with calcein solution, cells were analysed under a fluorescence microscope. We verified homogeneous growth in the culture chamber, without cell density gradients from inlet and outlet, and healthy morphology (Fig. 5a), confirming that the device is suitable for prolonged cell culture.

(a) HFF cells were seeded in the devices and stained with calcein in order to prove the long term biocompatibility of the device. (b) Time course of HFF cells stretching. (c) Percentage of area deformation profile at increasing pressures.
Stretching of the same cells was observed under the microscope set in contrast phase acquiring the video at 200 frames/s. In this experiment, the generated pressure difference was measured connecting a pressure manometer at the inlet of the serpentine circuit. In order to measure the total area deformation the acquired video was imported in the software ImageJ and single cell area was calculated for different time points (Fig. 5b). The so calculated area deformation was then correlated with the differential pressure measurements. Figure 5c shows the correlation between applied pressure difference and total area deformation. In the pressure range of 0.25–0.6 bar the relation is fairly linear, whereas at the beginning of the mechanical stimulation the linearity seems to fail, probably due to initial static friction. On the other hand the slope presents a plateau at about 20% area deformation indicating that the device has achieved the maximum possible deformation.
We next measured cell deformation using MCF10A cells, stably expressing membrane-bound EGFP and nuclear-localized mCherry. We injected cells in the device, let them adhere and then imaged in fluorescence to better visualize cellular membranes and thus cell deformation during stretch (Fig. 6a). In this case the frame acquisition rate was set at 2 frames/s. Total area deformation was measured as described previously, increasing the image contrast in order to highlight the cell contour and measuring the cell area at the beginning and at the end of the mechanical stimulation (Fig. 6b). Figure 6c shows the zoom-in of a single cell before and after the mechanical deformation and the comparison of the two cells area. The prototype used for this experiment is characterized by a wider central stretchable area then the previous one in order to increase the number of cells and consequently the statistical significance of the analysis. The calculated area deformation results 10.5 ± 1.1%.

(a) Integration of MCF10A cells stably expressing nuclear-bound mCherry and membrane-bound EGFP in the device. (b) EGFP expression of cells was highlighted using ImageJ software in order to better visualize the cell deformation between non stretched and stretched cells. (c) Zoom in of a single cell for cell surface comparison before and during stretching.
It is worth noting that fibroblasts were seeded in a device showing a central stretchable area half the width of that used with the MCF10A cells, namely 250 vs. 500 µm, so the difference in the maximum value of achievable stretch indicates that it is possible to tune the imposed mechanical strain both during the design of the experiment equipment and while performing the experiment.
The same prototype was used to analyse actin filaments remodelling occurring few minutes after stretch, by using a cell line stably expressing F-actin-bound EGFP. We designed the stretching experiment reported in Fig. 7a, in which we deformed cells from a resting position to 10% deformation (as measured in the previous experiment) and maintained the deformed status for a period of 10 min. Taking advantage of the microfluidic chamber where cells are cultured we also included the possibility to perturb cell microenvironment with Latrunculin B, a potent inhibitor of actin polymerization that disrupt filament organization, during the stretching and before starting images acquisition (i.e. between t0 and t1).

(a) Deformation profile as a function of time of U2OS cells, stably expressing Actin-bound EGFP. (b) Representative images of U2OS cells before stretching (black line) and during maximum deformation (red line). (c) Real time imaging of actin filaments during deformation as a function of time compared to non-stretching condition (where no changing of actin filaments can be observed) and in the presence of actin polymerization inhibitor Latrunculin B, used as positive control of actin remodelling. Arrows indicate changing of actin filaments observed in some cells during stretching.
Figure 7b shows that the focal plane is perfectly maintained during the imposed deformation and in the following recording period.
Actin filaments were imaged as a function of time during the imposed deformation in the presence or absence of Latrunculin B, as reported in Fig. 7c. If compared with no-stretched cells (Supplementary Video 1) a partial remodelling of actin filaments network can be observed in stretched cells (Supplementary video 2), mainly in terms of filaments length. Conversely, an almost complete destruction of actin filaments can be observed in few minutes if cells are exposed to 100 nM LatrunculinB, a potent actin polymerization inhibitor (Supplementary video 3).
These results demonstrate actin imaging can be easily performed with the developed device as a function of cell deformation and time. Ad hoc algorithms can be then used for a deeper analysis of the remodelling.2,30
Discussion
Mechano-transduction is continuing to increase its importance as a biological research field thanks to the growing knowledge of the key role of mechanical stimulations in regulating cell behaviour. The development of innovative research tools is therefore of the utmost importance to help unravel the complicated interconnection between mechano-sensing elements and biochemical pathways within the cell.
In this work we present a novel multi-layered microfluidic chip for cell culturing in a mechanically active environment. The device allows real-time high-resolution imaging of the experiment allowing studying the dynamic response of cultured cells to mechanical stimuli. Moreover, relaying entirely on microfluidic technologies, the soluble microenvironment can be tightly controlled and monitored.11
The design of the device was optimized using numerical simulation to identify the relation between operational parameters, such as membrane thickness, channels shape and applied pressure difference, and cell deformation. The device was further improved by subjecting the etched glass to a thermal treatment in order to reduce surface roughness and thus sliding of the elastic membrane.
The correct working of the serpentine shaped control channel was demonstrated and it was showed that the microfluidic device is fully biocompatible and is able to stretch cultured cells up to 20% total area deformation, showing a linear correlation between deformation and applied pressure difference.
The deformation imposed on the cultured cells can be easily tuned by varying the device design and the applied pressure difference. This allows exploring both physiological and pathological strain conditions and instantaneous responses can be easily analysed in more than one cells at the same time, thus increasing the readout significance.
A cell line previously used in the investigation of mechano-activated signalling pathways has been successfully seeded and stretched in the proposed device showing the promising ability of this technology to allow the real time visualization of the mechanical strain effect on cellular faith.
Taking advantage of the developed device we imaged actin filaments remodelling, by means of a fluorescent reporter cell line, in real time during the first minutes of stretching, observing partial remodelling of actin network, which could, in principle, be quantified by already developed algorithms.2,30
Overall, the novel developed device allows the unique combination of accurate cell mechanical stimulation, in line microscope analysis at high magnification while maintaining the focal plane and possibility to accurately control the soluble microenvironment thanks to automatic and precise medium delivery inside the microfluidic cell culture chamber. This will further support the increasing research in the field of mechano-transduction.
References
Ahmed, W. W., T. Wolfram, A. M. Goldyn, K. Bruellhoff, B. A. Rioja, M. Möller, J. P. Spatz, T. A. Saif, J. Groll, and R. Kemkemer. Myoblast morphology and organization on biochemically micro-patterned hydrogel coatings under cyclic mechanical strain. Biomaterials 31:250–258, 2010.
Alioscha-Perez, M., C. Benadiba, K. Goossens, S. Kasas, G. Dietler, R. Willaert, and H. Sahli. A Robust actin filaments image analysis framework. PLOS Comput. Biol. 12:e1005063, 2016.
Amaya, R., A. Pierides, and J. M. Tarbell. The interaction between fluid wall shear stress and solid circumferential strain affects endothelial gene expression. PLoS ONE 10:e0129952, 2015.
Antunes, M., T. Pereira, J. V. Cordeiro, L. Almeida, and A. Jacinto. Coordinated waves of actomyosin flow and apical cell constriction immediately after wounding. J. Cell Biol. 202:365–379, 2013.
Aragona, M., T. Panciera, A. Manfrin, S. Giulitti, F. Michielin, N. Elvassore, S. Dupont, and S. Piccolo. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154:1047–1059, 2013.
Balachandran, K., P. W. Alford, J. Wylie-Sears, J. A. Goss, A. Grosberg, J. Bischoff, E. Aikawa, R. A. Levine, and K. K. Parker. Cyclic strain induces dual-mode endothelial mesenchymal transformation of the cardiac valve. Proc. Natl. Acad. Sci. USA 108:19943–19948, 2011.
Chang, Y. J., C. J. Tsai, F. G. Tseng, T. J. Chen, and T. W. Wang. Micropatterned stretching system for the investigation of mechanical tension on neural stem cells behavior. Nanomed. Nanotechnol. Biol. Med. 9:345–355, 2013.
Chiquet, M., and M. Flück. Chapter 8 Early responses to mechanical stress: from signals at the cell surface to altered gene expression. In: Cell and Molecular Response to Stress, edited by K. B. Storey, and J. M. Storey. Amsterdam: Elsevier, 2001, pp. 97–110.
Dai, G., M. R. Kaazempur-Mofrad, S. Natarajan, Y. Zhang, S. Vaughn, B. R. Blackman, R. D. Kamm, G. García-Cardeña, and M. A. Gimbrone. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. 101:14871–14876, 2004.
Formigli, L., E. Meacci, C. Sassoli, R. Squecco, D. Nosi, F. Chellini, F. Naro, F. Francini, and S. Zecchi-Orlandini. Cytoskeleton/stretch-activated ion channel interaction regulates myogenic differentiation of skeletal myoblasts. J. Cell. Physiol. 211:296–306, 2007.
Giobbe, G. G., F. Michielin, C. Luni, S. Giulitti, S. Martewicz, S. Dupont, A. Floreani, and N. Elvassore. Functional differentiation of human pluripotent stem cells on a chip. Nat. Methods 12:637–640, 2015.
Giulitti, S., A. Zambon, F. Michielin, and N. Elvassore. Mechanotransduction through substrates engineering and microfluidic devices. Curr. Opin. Chem. Eng. 11:67–76, 2016.
Gudipaty, S. A., J. Lindblom, P. D. Loftus, M. J. Redd, K. Edes, C. F. Davey, V. Krishnegowda, and J. Rosenblatt. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 543:118–121, 2017.
Ishida, T., M. Takahashi, M. A. Corson, and B. C. Berk. Fluid shear stress-mediated signal transduction: how do endothelial cells transduce mechanical force into biological responses? Ann. N. Y. Acad. Sci. 811:12–23, 1997.
Johnston, A. P. W., J. Baker, M. D. Lisio, and G. Parise. Skeletal muscle myoblasts possess a stretch-responsive local angiotensin signalling system. J. Renin Angiotensin Aldosterone Syst. 12:75–84, 2011.
Kamble, H., M. J. Barton, M. Jun, S. Park, and N.-T. Nguyen. Cell stretching devices as research tools: engineering and biological considerations. Lab. Chip 16:3193–3203, 2016.
Kosmalska, A. J., L. Casares, A. Elosegui-Artola, J. J. Thottacherry, R. Moreno-Vicente, V. González-Tarragó, M. Á. del Pozo, S. Mayor, M. Arroyo, D. Navajas, X. Trepat, N. C. Gauthier, and P. Roca-Cusachs. Physical principles of membrane remodelling during cell mechanoadaptation. Nat. Commun. 6:7292, 2015.
Kumar, A., R. Murphy, P. Robinson, L. Wei, and A. M. Boriek. Cyclic mechanical strain inhibits skeletal myogenesis through activation of focal adhesion kinase, Rac-1 GTPase, and NF-κB transcription factor. FASEB J. 18:1524–1535, 2004.
Maul, T. M., D. W. Chew, A. Nieponice, and D. A. Vorp. Mechanical stimuli differentially control stem cell behavior: morphology, proliferation, and differentiation. Biomech. Model. Mechanobiol. 10:939–953, 2011.
McCain, M. L., and K. K. Parker. Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflüg. Arch. Eur. J. Physiol. 462:89, 2011.
Michielin, F., E. Serena, P. Pavan, and N. Elvassore. Microfluidic-assisted cyclic mechanical stimulation affects cellular membrane integrity in a human muscular dystrophy in vitro model. RSC Adv. 5:98429–98439, 2015.
Nakai, N., F. Kawano, Y. Oke, S. Nomura, T. Ohira, R. Fujita, and Y. Ohira. Mechanical stretch activates signaling events for protein translation initiation and elongation in C2C12 myoblasts. Mol. Cells 30:513–518, 2010.
Prosser, B. L., R. J. Khairallah, A. P. Ziman, C. W. Ward, and W. J. Lederer. X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 58:172–181, 2013.
Ruder, W. C., E. D. Pratt, N. Z. D. Brandy, D. A. LaVan, P. R. LeDuc, and J. F. Antaki. Calcium signaling is gated by a mechanical threshold in three-dimensional environments. Sci. Rep. 2:554, 2012.
Salameh, A., A. Wustmann, S. Karl, K. Blanke, D. Apel, D. Rojas-Gomez, H. Franke, F. W. Mohr, J. Janousek, and S. Dhein. Cyclic mechanical stretch induces cardiomyocyte orientation and polarization of the gap junction protein connexin43. Circ. Res. 106:1592–1602, 2010.
Shannon, E. K., A. Stevens, W. Edrington, Y. Zhao, A. K. Jayasinghe, A. Page-McCaw, and M. S. Hutson. Multiple mechanisms drive calcium signal dynamics around laser-induced epithelial wounds. Biophys. J. 113:1623–1635, 2017.
Shyy, J. Y.-J., and S. Chien. Role of integrins in cellular responses to mechanical stress and adhesion. Curr. Opin. Cell Biol. 9:707–713, 1997.
Suchyna, T. M., and F. Sachs. Mechanosensitive channel properties and membrane mechanics in mouse dystrophic myotubes. J. Physiol. 581:369–387, 2007.
Tulloch, N. L., V. Muskheli, M. V. Razumova, F. S. Korte, M. Regnier, K. D. Hauch, L. Pabon, H. Reinecke, and C. E. Murry. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ. Res. 109:47–59, 2011.
Vindin, H., L. Bischof, P. Gunning, and J. Stehn. Validation of an algorithm to quantify changes in Actin Cytoskeletal Organization. J. Biomol. Screen. 19:354–368, 2014.
Yeung, E. W., and D. G. Allen. Stretch-activated channels in stretch-induced muscle damage: role in muscular dystrophy. Clin. Exp. Pharmacol. Physiol. 31:551–556, 2004.
Acknowledgment
This research was supported by Progetti di Eccellenza CaRiPaRo, Oak Foundation Award (Grant #W1095/OCAY-14-191) and TRANSAC Progetto Strategico Universitá di Padova. This research was supported by the NIHR GOSH BRC. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Peter E. McHugh oversaw the review of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Prevedello, L., Michielin, F., Balcon, M. et al. A Novel Microfluidic Platform for Biomechano-Stimulations on a Chip. Ann Biomed Eng 47, 231–242 (2019). https://doi.org/10.1007/s10439-018-02121-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-018-02121-z