
Abstract
Mesenchymal stem cells (MSCs) are being studied extensively due to their potential as a therapeutic cell source for many load-bearing tissues. Compression of tissues and the subsequent deformation of cells are just one type physical strain MSCs will need to withstand in vivo. Mechanotransduction by MSCs and their mechanical properties are partially controlled by the cytoskeleton, including vimentin intermediate filaments (IFs). Vimentin IF deficiency has been tied to changes in mechanosensing and mechanical properties of cells in some cell types. However, how vimentin IFs contribute to MSC deformability has not been comprehensively studied. Investigating the role of vimentin IFs in MSC mechanosensing and mechanical properties will assist in functional understanding and development of MSC therapies. In this study, we examined vimentin IFs’ contribution to MSCs’ ability to deform under external deformation using RNA interference. Our results indicate that a deficient vimentin IF network decreases the deformability of MSCs, and that this may be caused by the remaining cytoskeletal network compensating for the vimentin IF network alteration. Our observations introduce another piece of information regarding how vimentin IFs are involved in the complex role the cytoskeleton plays in the mechanical properties of cells.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) have recently shown promise as a therapeutic cell source for the treatment of many diseases, including osteoarthritis (OA).1,15 However, osteoarthritic cartilage presents a challenge for therapeutic MSCs, injected systemically or implanted within a biomaterial scaffold, due to the abnormal biochemical and mechanical environment.8,11 One characteristic of the altered mechanical environment is regular compression that deforms the tissue and chondrocytes, eliciting extracellular matrix protein expression.2,12,17,21,32,35 Response to mechanical stresses is influenced by cellular mechanical properties. Changes in MSC mechanical properties have been found to be related to both their physical environment and differentiation potential.10,19,20,37 Mechanical properties and mechanotransduction are in part regulated by the cytoskeleton, consisting primarily of actin microfilaments, microtubules, and vimentin intermediate filaments (IFs) for cells of mesenchymal lineage.5,7,19,23,24,29,30,34,36 While their role in the pathogenesis of OA is still unknown, vimentin IFs have recently been found to be disrupted or dispersed in osteoarthritic chondrocytes.4,16 Notably, vimentin has also been shown to be downregulated in MSCs of OA patients,27 which raises questions about the potential efficacy of autologous stem cell therapies for treatment of OA.
Using a variety of techniques to decrease, disrupt, or collapse vimentin IFs, previous investigators have shown that the vimentin network is clearly involved in modulating the mechanical properties of cells. In fibroblasts, mutations resulting in vimentin deficiency have been linked to not only impaired migration, but also reduction of mechanical stability and stiffness of the cytoplasm.13,34 Further, in these cells, decreases in vimentin led to compromised ability for fibroblasts to contract collagen gels, which is critical for wound healing.7
Perinuclear collapse of vimentin networks in fibroblasts has also been induced using proteins, such as the oncogene simian virus 40 large T antigen26 or one variant of mutated desmin.25 Oncogene expression-dependent collapse of the vimentin network in fibroblasts caused an increase in cellular stiffness, which supports vimentin IFs association with tumor invasion and tumor cell stiffness.26 IF collapse caused by mutated desmin revealed a complex distribution of cellular stiffness with increased cellular stiffness in regions of the collapsed vimentin and a decrease in stiffness in the remaining vimentin-deficient cytoplasm.25
Collapse of the vimentin network has also been induced by pharmacological inhibitors such as withaferin A,9 calyculin A,3 and acrylamide.5,14,30,33 In non-adherent cell populations or cells suspended in hydrogels have revealed decreases in cellular mechanical properties with the use of pharmacological inhibitors. Specifically, in chondrocytes and chondrocyte-like cells, disruption of vimentin networks using acrylamide resulted in decreased elastic moduli and viscoelasticity, as measured by atomic force microscopy and micropipette aspiration, as well as a decrease in deformability.5,14,30,33 Likewise, in T lymphocytes and natural killer cells, disruption of vimentin IFs caused a decrease in cellular stiffness.3,9
Much of the research surrounding the role of vimentin IFs on cell mechanics and function has been conducted in fibroblasts by introducing the expression of non-natively expressed proteins or using pharmacological inhibitors, which may have off target effects that can influence the measurement of cellular stiffness. However, whether vimentin IFs similarly affect the biophysical properties of MSCs has not been established and an improved understanding of how IFs are involved in mechanosensing and mechanical properties of MSCs will be valuable for interpreting outcomes from stem cell therapies. In this study, we examine the relationship between MSCs’ capacity to deform under external compression and the involvement of vimentin IFs using shRNA mediated RNA interference (RNAi). The aim is to investigate the effect of a decreased vimentin IF network on MSC deformability independent of effects from cell-substrate adhesion and long culture times. Our results suggest that a decrease in vimentin IFs paradoxically reduces the deformability of MSCs, potentially due to changes in the manner by which actin microfilaments and microtubules organize and function to resist loads.
Materials and Methods
hMSC Cell Culture
For initial lentiviral construct screening experiments, hMSCs from Lonza (Walkersville, MD) were expanded per manufacturer’s instructions and used at passage 5 (P.5). For subsequent experiments, population doubling level (PDL) 9 bone marrow derived human mesenchymal stem cells (hMSCs) (RoosterBio; Frederick, MD) were expanded using RoosterBio Enriched Basal media supplemented with GTX Booster (RoosterBio) per manufacturer instructions and used at PDL 13–18 hMSCs (approximately 4–5 passages). All subsequent subculture for lentiviral transduction and experimentation was completed using hMSC growth media: high glucose DMEM containing 4 mM l-Glutamine (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U mL−1 Penicillin-Streptomycin (Gibco), 1% MEM non-essential amino acids (Gibco), and 4 mM l-Glutamine (Gibco). Complete media exchange was completed every 2–3 days and the cells were maintained at 5% CO2 and at 37 °C.
Lentivirus Design and Generation
52 nt shRNA sense-loop-antisense sequences were designed and selected from human vimentin [Gen Bank: NM_003380] mRNA using the shRNA Designer through Biosettia, Inc. Single strand oligonucleotides were annealed and these double stranded oligos were then ligated into an inducible lentiviral RNAi vector conveying resistance to blasticidin and a TetO-H1 promoter following manufacturer instructions. This inducible system only allows shRNA transcription to take place in the presence of tetracycline antibiotics, specifically doxycycline. The pLV-RNAi kit and pLV-Pack Packaging mix (Biosettia) were used to generate the shRNA constructs and package into replication-deficient lentivirus using HEK 293FT cells and Lipofectamine 2000. Two sequences were evaluated, listed in Table 1, and a control shRNA lentiviral vector targeting the LacZ gene was used (Biosettia). Virus-containing supernatants were collected 72 h post transfection and stored at −80 °C until use.
shRNA Transduction
We performed hMSC transduction with the shVim-vector for 24 h at a multiplicity of infection (MOI) of 15. Cells transduced with a shLacZ-vector and non-transduced cells were used as controls. Transduction was completed in the presence of 6 µg mL−1 hexadimethrine bromide (Polybrene) (Sigma) to assist with transduction efficiency. Titered viral concentrations for an MOI of 15 were determined through a Quanti-IT PicoGreen Assay (Invitrogen). Two days post-infection, pure populations were selected using 12 µg mL−1 Blasticidin for 4 days. Both shVim-hMSCs and shLacZ-hMSCs were cultured in the presence of 1 µg mL−1 doxycycline to induce RNAi. Cells were cultured for 7, 14, or 21 days on tissue culture plastic before being harvested to be assayed.
Western Blotting
To quantify levels of vimentin protein translation, cells were transduced and induction carried out for 7, 14, and 21 days. Cells were harvested and resuspended in a lysis buffer (50 mM HEPES, 150 mM sodium chloride, 1% Triton X-100, 1 mM EDTA, 10 mM Na-pyrophosphate, 10% glycerin) supplemented with a 1:100 concentration of protease inhibitor cocktail (Fisher Scientific). Protein concentrations were determined using a modified Lowry assay with a Folin-phenol color reaction detected by an ND-1000 spectophotometer (Nanodrop). After sample removal, the supernatant was mixed at a concentration of 1:1 with a loading buffer [13% (v/v) Tris–HCl, 20% (v/v) glycerol, 4.6% (w/v) SDS, 0.02% (w/v) bromophenol blue, 200 mM dithiothreitol]. Samples and a human vimentin protein positive control were subjected to SDS-PAGE using pre-cast Criterion Tris–HCl gels (BioRad). 293FT HEK cell lysate was used as a protein positive control for β-actin. Approximately 155 µg of protein from each sample was loaded into the Criterion Tris–HCl gels. After SDS-PAGE, proteins were electrophoretically transferred to a polyvinylidene fluoride membrane and detected using a rabbit IgG anti-human vimentin primary antibody (ThermoFisher) and Vectastain ABC-AmP for chromogenic detection. Detection of β-actin using a mouse IgG anti-human β-actin primary antibody was used as a loading control. Semi-quantitative analysis was completed using ImageJ (NIH) to determine band intensities and protein expression levels were determined relative to non-infected cells. For semi-quantitative analysis of vimentin protein expression levels, the top band of the cluster was used, as it aligns with the positive protein control.
Immunofluorescence Imaging
To visualize decrease in translated vimentin protein in 2D cultures, vimentin RNAi was induced for 7 and 14 days. Sham control (shLacZ) samples were performed in parallel. As an additional control, non-transduced cells were subjected to the RNAi-inducing agent (1 µg mL−1 doxycycline) for 14 days to determine its potential effects on cytoskeletal organization. Cells were fixed with 4% paraformaldehyde and permeabilized using 0.1% Triton X-100. Cells were labelled with either rabbit IgG anti-human vimentin primary antibody (ThermoFisher) or mouse IgG anti-human tubulin primary antibody (Santa Cruz) and visualized with biotinylated (anti-rabbit IgG or anti-mouse IgG) secondary antibodies (Vector) and fluorescein-labelled streptavidin (Vector). Actin filaments were then stained with Alexafluor 594 phalloidin (Invitrogen), and the nucleus counterstained with DAPI (Invitrogen). Fluorescence images were taken at ×100 magnification with an Olympus IX81 microscope.
To visualize the cytoskeleton antibodies in agarose gels, vimentin RNAi was induced for 14 days before being harvested. Sham control (shLacZ) samples were performed in parallel. Cells were then resuspended in 4% (w/v) agarose and pipetted into a 6 mm × 3 mm diameter mold, followed by overnight fixation in 4% paraformaldehyde. These were infiltrated with 30% sucrose, embedded in Tissue-Tek O.C.T compound (Sakura), and then stored at −80 °C until sectioning. Frozen sections (20 µm) were created using an HM550 series cryostat (Richard Allen Scientific). These sections were labelled with either rabbit IgG anti-human vimentin primary antibody (ThermoFisher) or rabbit IgG anti-human tubulin primary antibody (Abcam) and visualized with biotinylated anti-rabbit IgG secondary antibodies (Vector) and fluorescein-labelled streptavidin (Vector). In additional sections, actin filaments were stained with Alexafluor 488 Phalloidin (Invitrogen) and the nucleus stained using Slow Fade Gold anti-fade reagent with DAPI (Invitrogen). Confocal fluorescence images were taken at ×600 magnification with a Nipkow (spinning) disk-equipped Olympus IX81 microscope. Confocal Z-stacks (1 µm slices) of the entire cells were taken and projected into a single image for analysis. Fluorescence intensity of labelled proteins was quantified using Image J (NIH).22 Cells were manually traced and corrected total cell fluorescence intensity measurements per cell area were calculated using the following equation: corrected total cellular fluorescence (CTCF) = [integrated density − (area of selected cell × mean fluorescence of background reading)]/cell area (pixels). Data are shown as mean CTCF + SEM.
Cell Deformation
To measure cell deformation, after 14 days of inducing vimentin (and LacZ) RNAi cells were incubated with Cell Tracker Green CMFDA (Invitrogen) to stain the cell cytoplasm. Subsequently, 300–400k cells were resuspended in 2% (w/v) or 4% (w/v) agarose and pipetted into a 6 mm × 6 mm × 10 mm mold. After gels solidified, they were placed into a custom microscope-mounted micrometer-controlled deformation device.33 This process took at least two hours from time of trypsinization. Samples were then subjected to 0, 10, and 20% uniaxial bulk compressive strain. Fluorescence images of cells were generated at ×400 magnification and cell diameters in the loading direction and perpendicular to the loading direction were measured using ImageJ (NIH). Analysis was performed similar to a previously study.33 Aspect ratios (ARs) were calculated as cell diameter in the loading direction/cell diameter perpendicular to load, and the deformed population ARs were then normalized to the undeformed population ARs. Data are shown as mean normalized aspect ratio ± standard deviation.
Cytoskeletal Disruption
To determine the effect of microfilament and microtubule disruption on the shVim-hMSC and shLacZ-hMSC deformability, after 14–15 days of RNAi induction cells were incubated with CMFDA live cell tracker (Invitrogen). Afterward 300–400k cells were resuspended in 4% (w/v) agarose and pipetted into a 6 mm × 6 mm × 10 mm mold. Following encapsulation and prior to deformation, cell-agarose constructs were incubated with either 20 µM colchicine or 9.85 µM cytochalasin D for 3 h in 37 °C at 5% CO2 to disrupt microtubules or actin microfilaments, respectively.33 Agarose blocks were subjected to strain and the images analyzed, as described above. Data are shown as mean aspect ratio + standard deviation.
Statistical Analysis
Statistical analyses for all studies were performed using non-parametric Kruskal–Wallis tests followed by Mann–Whitney post hoc pairwise analyses using the statistical software SPSS. Statistical significance was set to α = 0.05.
Results
Inducible Lentiviral shRNA Mediated Knockdown of Vimentin Expression in hMSCs
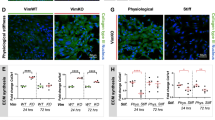
Initially, two shRNA vectors (Table 1), designed using different locations within the gene, were assessed for effectiveness in decreasing vimentin expression over 14 days in the presence of 1 µg mL−1 doxycycline, the highest recommended dose. Because shVim1 yielded greater RNAi than shVim2 (Fig. 1a), it was used for all subsequent experiments and is henceforth referred to as shVim. Cultures of shVim-transduced hMSCs exhibited a 40–60% decrease in vimentin expression, as shown by Western blot in the initial screen and in experiments to further characterize the vimentin knockdown by shVim (Figs. 1a and 1b). A decrease in vimentin protein was visible as seen by immunofluorescence in cells seeded on tissue culture plastic and in agarose hydrogels (Figs. 1c and 1d). Visually, we confirmed that 1 µg mL−1 doxycycline had negligible effect on the organization of vimentin, tubulin and F-actin in 2-D culture (Fig. 1e). Based on these results, it was determined that inducing RNAi for at least 14 days sufficiently knocked down vimentin protein levels, and this minimum induction period was used for all subsequent experiments.

Characterization of vimentin knockdown in hMSCs. (a) Two lentiviral vectors were screened using western blots and immunostaining on Day 14. In Western blots, ‘+Cntl’ is a purified vimentin protein positive control for Vim and a 293FT HEK cell lysate for B-Act. Scale bar 50 µm; (b) characterization of knockdown by Western blot on days 7, 14, and 21 of shRNA induction. ‘+Cntl’ is purified vimentin protein for Vim and 293FT HEK cell lysate for B-Act; (c) observation of vimentin knockdown by immunostaining on days 3, 7, and 14 of shRNA induction; vimentin (green), F-actin (red), nucleus (blue). Scale bar 50 µm; (d) observation of vimentin knockdown in agarose hydrogel; vimentin (green), nucleus (blue). Scale bar 50 µm. (e) Effect of 1 μg mL−1 doxycycline treatment on cytoskeletal proteins of control, non-transduced, hMSCs. Scale bar 50 µm.
Vimentin Knockdown Reduces hMSC Deformability
Knockdown of vimentin expression over 14 days resulted in decreased deformability of cells compared to both non-transduced hMSCs and shLacZ hMSCs in 4% agarose hydrogels. Compression of shVim-hMSCs yielded significantly higher normalized aspect ratios (Fig. 2a), or smaller deformations, compared to non-transduced hMSCs at 10% (p = 0.003) and 20% (p < 0.0005) strain (Fig. 2b), as well as compared to shLacZ-hMSCs at 20% strain (p < 0.0005). We found no significant difference between shLacZ-hMSCs and non-transduced hMSCs, indicating that lentiviral transduction did not significantly affect cellular deformability (10%, p = 0.528; 20%, p = 0.913; Fig. 2b). Interestingly, no significant difference was observed between any of the groups during deformation within the 2% agarose gels (10%, p = 0.182; 20% p = 0.093; Fig. 2c). Further, it was found that doxycycline treatment itself did not convey any resistance to the hMSCs (Fig. 2d). After 14 days of 1 µg mL−1 doxycycline treatment, no significant difference in deformation was observed between untreated and treated hMSCs in 4% agarose gels in any strain group (10%, p = 0.929; 20%, p = 0.383).

Cell deformation of vimentin-deficient hMSCs. Normalized aspect ratios of cells subjected to 0, 10, or 20% strain. (a) Deformation of control, non-transduced, hMSCs, shLacZ-hMSCs, shVim-hMSCs in 4% agarose hydrogels; (b) deformation of control, non-transduced, hMSCs, shLacZ-hMSCs, shVim-hMSCs in 2% agarose hydrogels; (c) deformation of control, non-transduced, hMSCs with or without treatment with 1 µg mL−1 doxycycline for 14 days. All data are expressed as mean aspect ratio ± SD. Asterisks represent statistically significant differences (p < 0.05).
Functional Role of Actin Filaments, but not Microtubules, is Altered by Vimentin Knockdown
To determine whether the reduced deformability of shVim-hMSCs was caused by changes in the actin or microtubule network, we exposed transduced cells seeded in 4% agarose to either cytochalasin D or colchicine, respectively. The non-transduced hMSCs sample group was not included in this experiment, because these cells were found to have no significant difference in deformability compared with shLacZ-hMSCs (Fig. 2). Comparisons in this experiment focused only on the effect of vimentin knockdown to the sham (shLacZ) control. After disruption of the microtubule network, shVim-hMSCs remained significantly less deformable at both 10% (p = 0.007) and 20% (p = 0.001) strain compared to shLacZ-hMSCs (Fig. 3a). In contrast, disrupting the actin microfilament network resulted in comparable cell deformations between shVim-hMSCs and shLacZ-hMSCs (Fig. 3b). Normalized aspect ratios were still slightly higher for shVim-hMSCs compared to shLacZ-hMSCs, but no longer significant at both 10% (p = 0.164) or 20% strain (p = 0.215).

Effect of cytoskeletal disruption on cell deformation. Normalized aspect ratios of shVim-hMSCs and shLacZ-hMSCs subjected to 0, 10, or 20% strain after chemical disruption of actin microfilaments or tubulin microtubules. (a) Deformation of shLacZ-hMSCs and shVim-hMSCs after actin microfilament disruption; (b) deformation of shLacZ-hMSCs and shVim-hMSCs after microtubule disruption. All data are expressed as mean aspect ratio ± SD. Asterisks represent statistically significant differences (p < 0.05).
Cytoskeletal Organization and Quantity in Agarose Embedded hMSCs
To further investigate the involvement of the actin or tubulin networks in the deformation of shRNA transduced cell populations, cytoskeletal protein content was semi-quantitatively determined from fluorescence microscopy of non-deformed cells. It was found that shVim-hMSCs and shLacZ-hMSCs did not have statistically significant differences in fluorescence intensities of F-actin staining (p = 0.267) (Fig. 4). However, the microtubule network fluorescence intensity was significantly lower in the shVim-hMSCs compared to the shLacZ-hMSCs (p = 0.01) (Fig. 4).

F-actin microfilament and tubulin microtubule fluorescence intensity in shVim-hMSCs and shLacZ-hMSCs. Fluorescent intensity measurements of shLacZ-hMSCs and shVim-hMSCs stained for F-actin and tubulin. All data are expressed as CTCF ± SEM. Asterisks represent statistically significant differences (p < 0.05). Scale bar 50 µm.
Discussion
Recently, studies have found vimentin IFs to be disrupted in chondrocytes and even in MSCs harvested from osteoarthritic bone marrow.4,16,27 Because mechanical loading is a strong regulator of cell behavior, we investigated how an altered vimentin network affects deformation of hMSCs during loading of agarose constructs. As a major component of the cytoskeleton, vimentin IFs are involved in the cellular response to mechanical loading and in modulating cellular mechanical properties. However, extracellular matrix and substrate stiffness also introduce changes in cell shape and cytoskeletal tension via adhesion complexes.20 Thus, the mechanical behavior of a cell is highly complex and context-dependent.
In this study, we focused on the deformation of MSCs in an experimental system that minimizes the ability for cells to form adhesion complexes with their surrounding microenvironment. As MSCs are not habitually unattached to extracellular matrix, this study provides a snapshot of the intrinsic deformability of undifferentiated MSCs. To prevent cell–matrix interactions, which would confound measurements of intrinsic deformability, we examined deformation of cells embedded in agarose hydrogels without allowing for extended culture time, as previously described.33
Contrary to expectations, our experiments showed that in 4% agarose hydrogels MSCs with decreased vimentin expression are more resistant to deformation compared to control cells. In an attempt to elucidate the mechanism behind this phenomenon, we additionally disrupted either actin microfilaments or microtubules. Although cells were generally more deformable with either treatment, only disruption of actin microfilaments eliminated the difference in deformability between shLacZ and shVim cells. That vimentin-deficient MSCs maintained a significantly greater resistance to deformation with microtubule disruption suggests a less prominent role for microtubules. Semi-quantitative measurement of the fluorescence intensity of immunostaining for F-actin and tubulin yielded further insight. Since actin fluorescence was unchanged, microfilament organization rather than quantity may be involved in the decreased deformability. The lower fluorescence intensity of the tubulin in shVim-hMSCs implies that the decrease in microtubules and organization of the actin microfilaments may work cooperatively to enhance resistance to cell deformation.
One obvious limitation of our RNAi approach is that vimentin IF expression is not completely ablated, unlike in fibroblasts isolated from vimentin null mice.7,34 On the other hand, our approach precludes any compensatory mechanisms that cells may develop physiologically in a knockout animal. Because vimentin continued to be expressed, albeit at a decreased level, we did not observe a complete collapse in the IF network with vimentin-silencing, as has been reported with acrylamide treatment.5,30 It is possible that the remaining vimentin network consists primarily of larger filaments, rather than the more diverse network of larger and smaller filaments that might support that strain normally. While large filaments were not observed in the immunostaining, Western blots did show a decrease in the smaller fragments in the knockdown cells compared to the control cells (Fig. 1b).
The increased resistance to deformation that we observed in the MSCs with decreased vimentin appears to contradict much of the literature in this area. Whole cell deformation experiments using chondrocytes and immune cells with chemically disrupted vimentin networks have resulted in mechanically less stiff and more deformable cells,3,5,9,14,30 though it is questionable how specific these treatments are for a given cytoskeletal target. Likewise in anchored vimentin-deficient fibroblasts, torsional loads applied via cell adhesions resulted in decreased stiffening or cytoplasmic rupture, suggesting that without vimentin, cells are mechanically unstable and unable to stiffen in response to load.7,34 It is not clear at this time how much our unexpected findings might be explained by the lack of cell–matrix attachment and the 3D hydrogel microenvironment of our experimental system.
Other studies, however, observed trends that are consistent with our results. One study reported a decrease in compressibility of acrylamide-treated chondrocytes.23 The authors postulated that vimentin IFs act as tensional elements preventing elongation orthogonal to the direction of compression, while microtubules prevent the compression of cells along the loading axis. Our data suggest that the actin microfilaments may play a more significant role in the resistance to deformation in the presence of a diminished vimentin network. Interestingly, dose-dependency of acrylamide treatment can also affect mechanical properties, implying a nonlinear relationship between the organization of vimentin and any change in cellular mechanical properties.30 Disruption of vimentin in chondrocytes using acrylamide was found to affect mechanical properties measured by micropipette aspiration only at high concentrations.30 Further, we have shown previously that chondrogenic hMSCs treated with this same high concentration of acrylamide trended toward increased deformability, but without statistically significant results.33 In this study, we did not observe a complete collapse of the vimentin network, and this could be why we see a dissimilar response to deformation in vimentin-deficient MSCs.
One critical parameter of this study is the choice of culture duration in the agarose hydrogel. Without significant culture time, cells would not be able to develop adhesion moieties that could subvert the deformation results. It has been observed that cytoskeletal proteins will undergo reorganization over chondrocyte culture time in agarose hydrogels over the timescale of days,18 implying that the cytoskeletal organization is dynamic over time. Here, we allowed a brief recovery after transfer to 3D culture in an attempt to capture an environment that simulates how vimentin may be involved in mechanosensing when hMSCs are first placed into a carrier biomaterial for therapy just prior to implantation. However, observations of cellular deformability at different stages in culture could provide more information about cell deformability and how microtubules and actin microfilaments reorganize to compensate for a less robust vimentin network over time. Further, longer culture periods would allow insight into changes in cellular phenotype due to 3-D culture, and changes in cellular behavior with the deposition of extracellular matrix.
The mechanical loads experienced by hMSCs in our experimental system are analogous to inclusions that deform within a loaded bulk porous material, which compounds the complexity of factors—beyond those associated with the cytoskeleton—that contribute to the cell deformation results. On a superficial level, the measurements that were made using 2 and 4% agarose can provide some insight into the balance of stiffness between cells and their surrounding material. In 2% agarose, cells deformed less across all groups than in 4% agarose, whose modulus is roughly five times that of 2%.6 It is possible that, due to the lower modulus of 2% agarose, cells were not subjected to sufficiently high compressive loads to resolve differences between shVim and shLacZ deformabilities. Delving deeper, some studies have shown differences in cytoskeletal organization with different agarose concentrations.28 Further, the non-linear mechanics of the cells might be distinct between shVim and shLacZ MSCs, where deformation may be similar under low load, but distinct at high load. Our previous work on chondrogenic hMSCs deficient in type VI collagen31 is one example of such behavior.
Some important aspects of cell deformation that we were not able to explore in this study include potential anisotropy of cell deformation, which would require more time consuming confocal imaging and 3D strain analysis of cells, as well as the possibility of discontinuities at the cell-gel interface due to differential stiffnesses that could interfere with analysis of cellular deformation. A more rigorous mechanical analysis of how cell deformability is governed in this complex system is certainly warranted. In this particular study, we chose a more straightforward approach to characterizing cell deformation in order to collect sufficient data for statistical comparisons between treatment groups.
While deformability measured by whole cell compression in 3D and stiffness measurements of anchored cells on a planar substrate yield different mechanical property relationships, it may be possible to relate the two sets of characteristics with further investigation. It has been speculated that the reduced mechanical stability observed in vimentin-negative fibroblasts may not necessarily be directly correlated with cellular flexibility, implying that their ability to withstand large deformations, e.g. migration through small pores, could be impaired.7 We have also observed impaired chemotactic migration in the vimentin knockdown MSCs and further found that robust vimentin networks may be required for migration through small pores (unpublished data). Further analysis of this phenomenon may shed light on the relationship between our observed deformation behavior in unanchored cells and cells experiencing mechanical stimuli due to adhesion and cytoskeletal remodeling during migration.
One variable of this study that has not been systematically studied in stem cells is MOI used for lentiviral transduction and its potential effects on cellular physiology. In order for us to achieve the desired knockdown of a gene as robustly expressed as vimentin, a relatively high MOI was required. Our previous studies using lentivirus-mediated RNAi in hMSCs had shown no detrimental effects on differentiation,31 but those prior experiments had used lower MOI. Though we did not observe any overt differences in morphology in any cells used for this present study, it is possible that other aspects of stem cell function may have been affected, independent from decreased vimentin. There has been some anecdotal evidence that MOI-dependent effects may be important.
This study reveals a unique relationship between vimentin IFs and MSCs’ capacity to deform due to external whole cell compression. Our observations suggest that deformability of MSCs is dependent on the robustness of the vimentin IF network in unanchored cells. Varying expression and organization of vimentin in healthy and diseased cells may affect the mechanical properties, and consequently the mechanotransduction, of these cells. Literature suggests that vimentin disruption or absence is present in OA chondrocytes and even MSCs from osteoarthritic patients, but it is not yet clear if this change is a symptom of the developing disease environment or an early actor in disease progression. In addition to examining vimentin’s role in the intrinsic properties of MSCs in agarose hydrogels, this study sheds initial light onto changes to mechanical properties that may occur to hMSCs due to an abrogated vimentin network that may be relevant in a cell therapy environment. Our observations introduce another variable and piece of information in understanding how IFs are involved in cellular mechanical properties.
Change history
02 January 2018
This erratum is to correct the following: (1) in the Western Blotting subsection under the Materials and Methods section, the concentration of protein from each sample loaded into Criterion Tris–HCl gels was incorrectly stated as 155 µg of protein. The correct value is 9.7 µg; (2) in Fig. 1b, the bar graph showed incorrect values for semi-quantitation of Western blots. Figure 1 has been updated with a corrected graph in Fig. 1b only.
References
Afizah, H., and J. H. P. Hui. Mesenchymal stem cell therapy for osteoarthritis. J. Clin. Orthop. Trauma 7:177–182, 2016.
Broom, N. D., and D. B. Myers. A study of the structural response of wet hyaline cartilage to various loading situations. Connect. Tissue Res. 7:227–237, 1980.
Brown, M. J., J. A. Hallam, E. Colucci-Guyon, and S. Shaw. Rigidity of circulating lymphocytes is primarily conferred by vimentin intermediate filaments. J. Immunol. 166:6640–6646, 2001.
Capín-Gutiérrez, N., P. Talamás-Rohana, A. González-Robles, C. Lavalle-Montalvo, and J. B. Kourí. Cytoskeleton disruption in chondrocytes from a rat osteoarthrosic (OA)-induced model: its potential role in OA pathogenesis. Histol. Histopathol. 19:1125–1132, 2004.
Chahine, N. O., C. Blanchette, C. B. Thomas, J. Lu, D. Haudenschild, and G. G. Loots. Effect of age and cytoskeletal elements on the indentation-dependent mechanical properties of chondrocytes. PLoS ONE 8:e61651, 2013.
Chen, Q., B. Suki, and K.-N. An. Dynamic mechanical properties of agarose gels modeled by a fractional derivative model. J. Biomech. Eng. 126:666–671, 2004.
Eckes, B., D. Dogic, E. Colucci-Guyon, N. Wang, A. Maniotis, D. Ingber, A. Merckling, F. Langa, M. Aumailley, A. Delouvée, et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 111:1897–1907, 1998.
Fukui, N., C. R. Purple, and L. J. Sandell. Cell biology of osteoarthritis: the chondrocyte’s response to injury. Curr. Rheumatol. Rep. 3:496–505, 2001.
Gladilin, E., P. Gonzalez, and R. Eils. Dissecting the contribution of actin and vimentin intermediate filaments to mechanical phenotype of suspended cells using high-throughput deformability measurements and computational modeling. J. Biomech. 47:2598–2605, 2014.
González-Cruz, R. D., V. C. Fonseca, and E. M. Darling. Cellular mechanical properties reflect the differentiation potential of adipose-derived mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 109:E1523–E1529, 2012.
Guilak, F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 25:815–823, 2011.
Guilak, F., A. Ratcliffe, and V. C. Mow. Chondrocyte deformation and local tissue strain in articular cartilage: a confocal microscopy study. J. Orthop. Res. 13:410–421, 1995.
Guo, M., A. J. Ehrlicher, S. Mahammad, H. Fabich, M. H. Jensen, J. R. Moore, J. J. Fredberg, R. D. Goldman, and D. A. Weitz. The role of vimentin intermediate filaments in cortical and cytoplasmic mechanics. Biophys. J. 105:1562–1568, 2013.
Haudenschild, D. R., J. Chen, N. Pang, N. Steklov, S. P. Grogan, M. K. Lotz, and D. D. D’Lima. Vimentin contributes to changes in chondrocyte stiffness in osteoarthritis. J. Orthop. Res. 29:20–25, 2011.
Jo, C. H., Y. G. Lee, W. H. Shin, H. Kim, J. W. Chai, E. C. Jeong, J. E. Kim, H. Shim, J. S. Shin, I. S. Shin, J. C. Ra, S. Oh, and K. S. Yoon. Intra-articular injection of mesenchymal stem cells for the treatment of osteoarthritis of the knee: a proof-of-concept clinical trial. Stem Cells 32:1254–1266, 2014.
Lambrecht, S., G. Verbruggen, P. C. M. Verdonk, D. Elewaut, and D. Deforce. Differential proteome analysis of normal and osteoarthritic chondrocytes reveals distortion of vimentin network in osteoarthritis. Osteoarthritis Cartilage 16:163–173, 2008.
Lee, D. A., and D. L. Bader. The development and characterization of an in vitro system to study strain-induced cell deformation in isolated chondrocytes. Vitro Cell. Dev. Biol. Anim. 31:828–835, 1995.
Lee, D. A., M. M. Knight, J. F. Bolton, B. D. Idowu, M. V. Kayser, and D. L. Bader. Chondrocyte deformation within compressed agarose constructs at the cellular and sub-cellular levels. J. Biomech. 33:81–95, 2000.
Lee, J.-H., H.-K. Park, and K. S. Kim. Intrinsic and extrinsic mechanical properties related to the differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 473:752–757, 2016.
Mathieu, P. S., and E. G. Loboa. Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways. Tissue Eng. Part B Rev. 18:436–444, 2012.
Mauck, R. L., B. A. Byers, X. Yuan, and R. S. Tuan. Regulation of cartilaginous ECM gene transcription by chondrocytes and MSCs in 3D culture in response to dynamic loading. Biomech. Model. Mechanobiol. 6:113–125, 2006.
McCloy, R. A., S. Rogers, C. E. Caldon, T. Lorca, A. Castro, and A. Burgess. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13:1400–1412, 2014.
Ofek, G., D. C. Wiltz, and K. A. Athanasiou. Contribution of the cytoskeleton to the compressive properties and recovery behavior of single cells. Biophys. J. 97:1873–1882, 2009.
Pan, W., E. Petersen, N. Cai, G. Ma, J. R. Lee, Z. Feng, K. Liao, and K. W. Leong. Viscoelastic properties of human mesenchymal stem cells, 2005. doi:10.1109/IEMBS.2005.1615559.
Plodinec, M., M. Loparic, R. Suetterlin, H. Herrmann, U. Aebi, and C.-A. Schoenenberger. The nanomechanical properties of rat fibroblasts are modulated by interfering with the vimentin intermediate filament system. J. Struct. Biol. 174:476–484, 2011.
Rathje, L.-S. Z., N. Nordgren, T. Pettersson, D. Rönnlund, J. Widengren, P. Aspenström, and A. K. B. Gad. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA 111:1515–1520, 2014.
Rollín, R., F. Marco, E. Camafeita, E. Calvo, L. López-Durán, J. Á. Jover, J. A. López, and B. Fernández-Gutiérrez. Differential proteome of bone marrow mesenchymal stem cells from osteoarthritis patients. Osteoarthr. Cartil. 16:929–935, 2008.
Steward, A. J., D. R. Wagner, and D. J. Kelly. The pericellular environment regulates cytoskeletal development and the differentiation of mesenchymal stem cells and determines their response to hydrostatic pressure. Eur. Cell Mater. 25:167–178, 2013.
Titushkin, I. A., and M. R. Cho. Controlling cellular biomechanics of human mesenchymal stem cells, 2009. doi:10.1109/IEMBS.2009.5333949.
Trickey, W. R., T. P. Vail, and F. Guilak. The role of the cytoskeleton in the viscoelastic properties of human articular chondrocytes. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 22:131–139, 2004.
Twomey, J. D., P. I. Thakore, D. A. Hartman, E. G. H. Myers, and A. H. Hsieh. Roles of type VI collagen and decorin in human mesenchymal stem cell biophysics during chondrogenic differentiation. Eur. Cell. Mater. 27:237–250, 2014.
Urban, J. P. The chondrocyte: a cell under pressure. Br. J. Rheumatol. 33:901–908, 1994.
Vigfúsdóttir, Á. T., C. Pasrija, P. I. Thakore, R. B. Schmidt, and A. H. Hsieh. Role of pericellular matrix in mesenchymal stem cell deformation during chondrogenic differentiation. Cell. Mol. Bioeng. 3:387–397, 2010.
Wang, N., and D. Stamenović. Contribution of intermediate filaments to cell stiffness, stiffening, and growth. Am. J. Physiol. Cell Physiol. C279:C188–194, 2000.
Wu, J. Z., W. Herzog, and M. Epstein. Modelling of location- and time-dependent deformation of chondrocytes during cartilage loading. J. Biomech. 32:563–572, 1999.
Yourek, G., M. A. Hussain, and J. J. Mao. Cytoskeletal changes of mesenchymal stem cells during differentiation. ASAIO J. (Am. Soc. Artif. Intern. Organs 1992) 53:219–228, 2007.
Yu, H., C. Y. Tay, W. S. Leong, S. C. W. Tan, K. Liao, and L. P. Tan. Mechanical behavior of human mesenchymal stem cells during adipogenic and osteogenic differentiation. Biochem. Biophys. Res. Commun. 393:150–155, 2010.
Acknowledgments
This work was supported by the National Science Foundation (CMMI 1563721, DRW; CBET 0845754, AHH).
Conflict of interest
No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Eric M. Darling oversaw the review of this article.
A correction to this article is available online at https://doi.org/10.1007/s10439-017-1975-5.
Rights and permissions
About this article

Cite this article
Sharma, P., Bolten, Z.T., Wagner, D.R. et al. Deformability of Human Mesenchymal Stem Cells Is Dependent on Vimentin Intermediate Filaments. Ann Biomed Eng 45, 1365–1374 (2017). https://doi.org/10.1007/s10439-016-1787-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-016-1787-z