
Abstract
Bioactive glass (BG) based scaffolds (45S5 BG composition) were developed by the replica technique using natural marine sponges as sacrificial templates. The resulting scaffolds were characterized by superior mechanical properties (compression strength up to 4 MPa) compared to conventional BG scaffolds prepared using polyurethane (PU) packaging foam as a template. This result was ascribed to a reduction of the total scaffold porosity without affecting the pore interconnectivity (>99%). It was demonstrated that the reduction of total porosity did not affect the bioactivity of the BG-based scaffolds, tested by immersion of scaffolds in simulated body fluid (SBF). After 1 day of immersion in SBF, a homogeneous CaP deposit on the surface of the scaffolds was formed, which evolved over time into carbonate hydroxyapatite (HCA). Moreover, the enhanced mechanical properties of these scaffolds were constant over time in SBF; after an initial reduction of the maximum compressive strength upon 7 days of immersion in SBF (to 1.2 ± 0.2 MPa), the strength values remained almost constant and higher than those of BG-based scaffolds prepared using PU foam (<0.05 MPa). Preliminary cell culture tests with Saos-2 osteoblast cell line, namely direct and indirect tests, demonstrated that no toxic residues remained from the natural marine sponge templates and that cells were able to proliferate on the scaffold surfaces.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bone is a specialized, connective, dynamic, and highly vascularized tissue able to remodel during the entire lifetime of mammals.28 Bone plays a key role in human physiology including locomotion and protection of vital organs imparting the skeleton with adequate load-bearing capability. Bone is also involved in haematopoiesis and in homeostasis through its storage of calcium and phosphorous ions.25 The high regenerative capability of bone means that the majority of fractures will usually heal without need for major interventions. Despite this ability, large bone defects lack support for an orchestrated regeneration and often require surgical intervention. Diseases along with traumatic injuries and primary tumor resection lead to large bone defects and/or voids25 needing a substitutionary material to fill the defect. The present “gold standard” is to harvest “donor” bone from a non-load-bearing site and transplant it into the defect site of the same patient.28 The main limitations of this approach are the additional operating time, required healing of both donor and implant sites, as well the pain, increased risk of infections2 and prolonged hospitalization and rehabilitation periods.2,4,25,28
Tissue engineering (TE) represents an alternative to using autograft tissue.4 As is frequently reported,2,28 tissue engineering is a multidisciplinary field whose final goal is the development of biological substitutes that restore tissue function. TE approaches combine biomaterials in the form of porous structures (termed “scaffolds”), cells and growth factors in order to create living, physiological, three dimensional tissues.11 One of the current challenges in TE is the design of scaffolds that can match both the mechanical and biological properties of the bone tissue matrix and can support the osteogenic potential and neovascularisation of large tissue constructs.11
It is now accepted that no foreign material placed within the human body is completely bioinert.14,15 In fact, when a synthetic material is placed in the physiological environment, tissues react in different ways depending on the material type. There are three terms used to classify biomaterials depending on their tissue response: almost bioinert, bioactive and bioresorbable.15 “A Bioactive material is defined as a material that elicits a specific biological response at the interface of the material, which results in the formation of a bond between the tissue and the material”.14 This definition was put forward by Hench, who discovered in early studies in the 1960s that certain compositions of silicate glasses in the system SiO2–CaO–Na2O–P2O5 are able to form a bond with both soft and hard tissues.13,14 Such group of surface reactive glasses were termed bioactive glasses. Indeed, when a bioactive glass (BG) is brought into contact with biological fluids a layer of carbonated hydroxyapatite (HCA), similar to the mineral phase of bone, is deposited on the surface and the release of dissolution products from the surface stimulates gene expression promoting osteoinduction and neo-angiogenesis.11,12,16,17,30 In the context of bioactive glasses intended for bone regeneration, it is now generally recognized that the key mechanisms leading to osteoinduction15,17 are related to the controlled release of ionic dissolution products from degrading bioactive glasses, which considers specially critical concentration of soluble silica and calcium ions, at the rate needed for cell proliferation and differentiation.17,30 Bioactive glass of 45S5 composition (45SiO2–24.5Na2O–24.5CaO–6P2O5 in wt%) was the first BG developed and today it is still the most frequently investigated BG for bone defect repair.18 45S5 BG has been used in several clinical applications in bulk form and as particulate for bone grafting and for the prevention of dental hypersensitivity. Jones18 has recently summarized the different traditional BG applications in orthopedic, dentistry and bone regeneration whilst emerging applications in soft tissue engineering have been reviewed elsewhere.22
Porous 3D BG based scaffolds are not yet available for clinical applications. The main difficulty is the fabrication of a scaffold that has, at the same time, high porosity, sufficient mechanical stability and fracture strength similar to that of natural bone. One approach is the manufacture of BG based scaffolds with improved mechanical properties due to a reduction of the volumetric porosity without affecting the pore interconnectivity and maintaining adequate pore size for new tissue ingrowth and vascularization. The first 3D BG scaffolds based on 45S5 BG composition were introduced in 2006 by Chen et al., 9 who used a standard foam replica technique with polyurethane (PU) foam as template to fabricate scaffolds of porosity >90%. Despite their high bioactivity9 and vascularization potential,3 these scaffolds do not have sufficient mechanical strength to be used in load bearing bone sites. More recently, 3D BG based scaffolds have been produced using a powder metallurgy approach1,6 and by applying the foam replica method to marine natural sponges.5,7 The foams produced with both these techniques were structurally robust showing compressive strength values comparable with those of cancellous bone (2–12 MPa) and exhibiting high pore connectivity1,5–7 and stable mechanical properties also after 28 days in simulated body fluid (SBF).6 These results are similar to those obtained on coralline HCA scaffolds20 in terms of mechanical properties and total porosity, however the BG-based scaffolds have the advantage to be biodegradable11 and also the ability to release biologically active ions to promote bone formation and angiogenesis.12,17 To date, however, marine sponge derived BG scaffolds have not been extensively investigated in terms of bioactivity, biodegradability and cell biology response. Therefore, the present work provides an in depth study on a novel family of BG scaffolds based on natural marine sponges. In particular, the effect of pore structure on the bioactivity and mechanical stability in SBF of BG-based scaffolds produced via a replication method of two natural marine sponges, namely Spongia Agaricina and Spongia Lamella, was investigated. Results of preliminary cell culture studies using Saos-2 osteoblasts on the scaffolds are also presented. In a previous work it was demonstrated that these sponges, thanks to the millenarian evolution for water filtration, could be a potential candidate for the production of bone tissue engineered scaffolds due to their interconnected pore architecture and suitable mechanical properties.5,7,10 A comparison between these novel scaffolds and those produced using PU foam as template9 is also presented.
Materials and Methods
Scaffolds Preparation and Characterisation
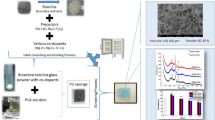
The starting material was melt-derived 45S5 BG powder (nominal mean particle size 2 µm). The sacrificial templates used for scaffolds production were PU packaging foams (45 ppi) (Eurofoam Deutschland GmbH Schaumstoffe, Germany), marine sponges “Spongia Agaricina” (SA) and “Spongia Lamella” (SL) harvested respectively in Indo-Pacific Ocean (Pure Sponges, UK) and Mediterranean Sea (Hygan Products Limites, UK) belonging to the “Elephant Ears” family. Harvesting of all natural sponges was performed in an environmentally-friendly manner, as specified by the suppliers. BG scaffolds were produced by the replica technique, according to the method described by Chen et al. 9 Briefly, a BG slurry was prepared by dissolving polyvinyl alcohol (PVA) in deionized water at 80 °C for 1 h (ratio 0.01 mol L−1). Then BG powder was added to PVA-water solution up to a concentration of 40 wt%. The sacrificial templates, cut into cylindrical shape, were immersed in the slurry for 10 min. The foams were then retrieved from the suspension and the extra slurry was manually squeezed out. The samples were then dried in air at room temperature. This procedure was repeated two or three times to increase the coating thickness and consequently the mechanical properties of the scaffolds. After the second and third coatings, the superfluous slurry was removed using compressed air, as reported in a previous work.7 The PU foams and SL sponges needed to be immersed in the slurry three times, while for SA sponge only two impregnations were sufficient to obtain the scaffolds. After drying, the samples were submitted to a heat treatment to remove the sacrificial sponges and to densify the structure. The sacrificial sponge burnout and the sintering conditions were: 400 and 1050 °C/1 h, respectively, with a heating rate of 2 °C min−1 and a cooling rate of 5 °C min−1. The process is summarized in Fig. 1.

Schematic diagram showing the preparation of 45S5 BG based scaffolds via replica method: after the preparation of the green body the sacrificial templates were burned out and the BG sintered by heat treatment in air.9
The resulting scaffolds were immersed in SBF for up to 28 days in order to evaluate their bioactivity and mechanical stability in a physiological like environment. SBF was prepared as reported by Kokubo and Takadama.19 For SBF preparation, 8.035 g L−1 NaCl, 0.355 g L−1 NaHCO3, 0.225 g L−1 KCl, 0.231 g L−1 K2HPO4 (3H2O), 0.311 g L−1 MgCl2 (6H2O), 0.292 g L−1 CaCl2 and 0.072 g L−1 Na2SO4 were dissolved in deionised water and buffered at pH 7.4 at 36.5 °C with 6.118 g L−1 tris(hydroxymethyl) aminomethane ((CH2OH)3CNH2) and 1 M HCl. Cylindrical BG foams were immersed in SBF with a 1.5 g L−1 ratio which has been proposed in previous studies.8,21 The specimens were kept in a polypropylene container at 37 °C in incubator on an oscillating tray for up to 28 days. The solution was renewed every 48 h in order to better mimic in vivo conditions.9 At each time point, foams were collected, rinsed three times with deionised water and dried at room temperature for 3 days. The microstructural changes were investigated by means of scanning electron microscopy SEM–EDS (Auriga 0750 from ZEISS). The 3D structure of the foams was observed using µ-CT scanner (Skyscan 1147 Micro-CT, Bruker). The pore interconnectivity was evaluated using CTan image analysis software. The foams were also scanned using a high resolution µ-CT scanner (Skyscan 1172 Micro-CT, Bruker, Belgium). Scanning was carried out at a resolution of 5.15 µm/pixel over 360° rotation with a step of 0.4°. Cell size was calculated using a volume-based approach with the software CT-Analyser (1.1.13, Skyscan B.V., Kontich, Belgium).
The density of the foams, ρ foams, was determined using the mass and dimensions of the sintered cylinders. The porosity, p, was calculated with the formula:
where ρ solid = 2.7 g cm−3 is the theoretical density of 45S5 BG, not considering a possible change of density due to the crystallization of the material during the thermal treatment or after the HCA precipitation.16 The nucleation of HCA was assessed by FTIR (Nicolet 6700, Thermo Scientific Germany), using KBr pellets and 32 scans at a resolution of 4 cm−1, which were repeated over the wavenumber range of 4000–400 cm−1.
The unchanged SBF was kept for the analysis of the concentration of Ca, P and Si ions through inductively coupled plasma optical emission spectroscopy ICP-OES (Optima 8300 Perkin Elmer) and for pH analysis. The percentage of the glass dissolution was calculated based on ICP results according to the following formula:
where [ion]ICP is the concentration measured in solution by ICP analysis at a given time-point and [ion]BG is the theoretical concentration in the BG foams.
The compression strength of BG foams, in as-fabricated state and soaked for 7, 14, 21 and 28 days in SBF, was measured using a Zwick Z050 mechanical tester at a crosshead speed of 1 mm min−1, with load cells of 50 N and 1 kN. The load was applied until the densification of the samples started to occur. The compressive strength was defined as the maximum stress of the linear elastic part of the stress–strain curve.
Indirect Cell Culture Tests
Indirect cell culture tests were performed following the general guidance set by the International Standardization Organization (ISO 10993-5:2009). The extracting medium was prepared by placing the not-preconditioned scaffolds, sterilized at 120 °C for 1 h, in cell culture medium for 1 day at 37 °C under 5% CO2 in a culture incubator. The total amount of cell culture medium per single scaffolds was a function of the scaffold weight (because the specific surface area of scaffolds was not available). The mass/extraction volume recommended by ISO standard is 0.1 g mL−1. After 1 day the cell culture medium in contact with the scaffolds was collected and used for the test. Saos-2 human osteoblast-like cells were seeded in fresh and conditioned media. The conditioned medium was also diluted by 50, 10 and 1% and used for cytotoxicity evaluation. The cell culture was conducted for 24 h as recommended by ISO standard. The cell culture medium was McCoy cell culture medium (Sigma M4892, NaHCO3 free) containing 1% vol. sodium pyruvate (Sigma S8736), 1% vol. penicillin-streptomycine solution (Sigma P0781) and 15% vol. fetal bovine serum (Sigma F7524). The viability of the cells was tested with Alamar Blue assay, measuring the optical fluorescence at an excitation wavelength of 570 nm and emission wavelength of 590 nm. The viability of the cells cultured with fresh and aged medium was used as control.
Direct Cell Culture Test
In order to evaluate the cell behaviour once in direct contact with the BG scaffolds, a direct cell culture test was carried out. Every scaffold was pre-conditioned in 2 mL of cell culture medium with 30 mM HEPES in an incubator (37 °C, 5% CO2) until the pH was lower than 8. Previous studies have shown that cells better proliferate on BG scaffolds after a preconditioning treatment in order to avoid the rapid increase of pH over non-physiological values.29 Due to this pre-conditioning step, it was considered unlikely that scaffold degradation would affect the cell attachment during cell culture tests. In fact the higher weight loss of the scaffolds took place during the first day of immersion. In the present study the medium was changed every day and the pH was continuously monitored. After 1 week the samples were washed with PBS to remove the remaining medium from the inner core of the scaffolds, immersed for 1 h in fresh cell culture medium and dried. Saos-2 cells were seeded at a density of 5 × 105 cell cm−2 in 2 mL of fresh culture medium and cultured under an atmosphere of 5% CO2 at 37 °C up to 14 days. The cell culture medium used for the direct cell culture was the same used for the indirect cell test but without HEPES in order not to affect the cell viability and proliferation. The viability of cells was evaluated using the mean of the Alamar Blue assay. The culture medium was renewed every 2 days.
The starting SA sponges are characterized by an inhalant and an exhalant surface, referring to the direction of water flow in the sponges in their natural habitat.26 For the resulting BG-SA scaffolds it was possible to distinguish the inhalant and exhalant surfaces, as reported previously.5,7 For this reason, cells were seeded on both surfaces in order to verify if their viability was affected by the different surface structure of the scaffolds.
Statistical Analysis
Significant differences in cell culture tests were determined using a one-way analysis of variance (ANOVA) with a Tukey multiple comparison post-test. Differences were considered significant at p values <0.05.
Results
Weight and Porosity Variation
The as-sintered scaffolds BG-SA, BG-SL and BG-PU were characterized, respectively, by porosities of 68.0 ± 0.2, 76 ± 3 and 93.0 ± 0.3% and pore sizes of 215 ± 3, 265 ± 20 and 670 ± 70 µm, respectively, as reported in a previous work.7 The total porosities were determined by measurements of mass and dimensions of samples and applying Eq. (1). After the immersion in SBF a decrease of the weight (Fig. 2a) and an increase of the total porosity (Fig. 2b) were observed for both BG-SA and BG-SL samples. Both types of scaffolds prepared with natural marine sponges as the sacrificial template lost between 40 and the 50% of their starting weight upon 28 days in SBF. The main weight change was observed in the first week of the test. The porosity of the BG-SA samples, at the end of the test, increased up to 80% and the porosity of the BG-SL samples increased up to 88%. For the BG-PU scaffolds it was not possible to analyze the weight and porosity variation because the samples were almost completely degraded due to their high dissolution in SBF and fragility with repeat handling.

(a) Weight and (b) porosity variation of BG-SA and BG-SL scaffolds after different immersion times in SBF for up to 28 days. The standard deviation values of the data for BG-SL in (b) were too low to be visible.
Mechanical Stability
By testing ten samples in as-fabricated conditions the resulting compressive strengths values were determined as follows: 1.8 ± 0.3 MPa for BG-SL, 4.0 ± 0.4 MPa for BG-SA and <0.05 MPa for BG-PU (the measurements were below the detection limit of the equipment) scaffolds, as reported in a previous work.7 BG-PU scaffolds were too fragile to be handled and it was not possible to perform the compression strength test on these samples. This was probably a consequence of the reduction of weight and increase of the volumetric porosity, after soaking in SBF. On the other hand, even after 28 days in SBF, it was still possible to handle BG-SA and BG-SL scaffolds without damaging them. For BG-SA and BG-SL scaffolds, the highest loss of mechanical strength took place during the first 7 days of immersion when a reduction of 50% from the starting maximum compressive strength values occurred. At the end of the test, the resulting compressive strength values were 1.2 ± 0.2 MPa for BG-SA and 0.23 ± 0.01 MPa for BG-SL samples (Fig. 3a). In both cases the values were higher than the maximum compressive strength of as-sintered BG-PU samples.

Variation of the mechanical properties of BG-SA and BG-SL scaffolds after different immersion times in SBF up to 28 days.
Ion Release and pH Variation
The pH variation and the changes of ion concentration in SBF during the first hours of immersion are reported in Fig. 4. Positive values of ion release imply an increase of the ion concentration in SBF, while negative values refer to a depletion of the ion concentration in SBF due to the immersion of the samples. Scaffold dissolution took place immediately after the first hours of immersion, as shown by the rapid increase in pH and the continuous ion release. After 3 days of immersion in SBF around 40 mg L−1 of Si species was released from both the BG-SA and BG-SL samples. The corresponding Si release from BG-PU was higher, at almost 61 mg L−1. The Ca concentration increased significantly after 6 h, reaching around 10 and 25 mg L−1, respectively for natural sponges (both BG-SA and BG-SL) and BG-PU samples. The P concentration in SBF decreased over time, indicating that a P-containing material was deposited on the surface of the scaffolds. No pH equilibrium was reached even after 2 days of testing, suggesting a continuous dissolution of the scaffolds.

Concentration of (a) Si, (b) Ca and (c) P ions released from BG scaffolds during the first hours of immersion in SBF up to 3 days; (d) pH variation in the first 48 h of immersion in SBF.
Surface Modification Analysis: SEM and FTIR
SEM micrographs of foams, before and after immersion in SBF for 1, 3, 7, 14 and 28 days are reported in Fig. 5. Already after 1 day of immersion a deposit was seen to form on the entire surface of the scaffolds. After 3 days of immersion (Figs. 5c, 5i, and 5o), the deposit increased in thickness and the cauliflower-like structure (typical of HCA)5 was identified. At 28 days of immersion in SBF, the deposit continued to grow and the needle-like structure of the HCA phase was clearly revealed. The same results were obtained for scaffolds prepared using PU foams and both natural marine sponges. No perceivable differences were identified in terms of HCA deposit rate and evolution of the deposit. It was observed that in all cases, the struts of the scaffolds were completely covered by HCA, as documented in Figs. 5(f), 5(l), and 5(r).

SEM micrographs of BG-SA (a–f), BG-SL (g–l) and BG-PU (m–r) scaffolds at different immersion times in SBF (up to 28 days) showing evolution of HCA deposit.
Figure 6 summarizes FTIR spectra of the samples before and after immersion in SBF for up to 28 days. In the case of the reference 45S5 BG, the FTIR spectra at room temperature presented the characteristic peaks of the non-bridging oxygen stretching mode of Si–O located at 950 cm−1 23 After 14 days, characteristic peaks ascribed to the HCA layer formation appeared as a doublet at around 600 cm−1, corresponding to the bending mode of P–O (crystalline phosphate).23 Moreover, the observation of P–O stretching at around 1000 cm−1, where the band became narrow, suggested the presence of HCA.23 The spectra also showed the narrowing of the band at around 800 cm−1 corresponding to the bending mode of C–O and finally the manifestation of the stretching mode of C–O at around 1400 cm−1 23 The SEM micrograph in Fig. 6 shows a well-developed HCA layer on the BG-SA scaffold surface after 28 days in SBF.

FTIR analysis results on BG-SA (a), BG-SL (b) and BG-PU (c) scaffolds before and after immersion in SBF, and SEM micrograph (d) showing the HCA needle like structure formed on the BG-SA surface after immersion in SBF for 28 days.
Micro-CT Analysis
Analysis of the 3D structure of the present scaffolds by micro-CT was carried out in order to gain insight on how immersion in a physiological-like environment could influence the scaffold porosity. In Fig. 7, typical 2D reconstruction images of the BG scaffolds immersed in SBF for up to 28 days are reported. For the samples obtained from natural marine sponges, it is observed that after 7 days the material became less dense at its edge (reduction in brightness) due to the dissolution process starting from the external surface of the foams. After 14 days, this less dense area expanded deeper into the specimens. The scans at 28 days revealed the formation of an external layer of a more dense material, likely related to the HCA layer observed by SEM (Fig. 5). The white arrows in Fig. 7(a) indicate the thicker layer of HCA, grown after 28 days in SBF. For the BG-PU scaffolds it was possible to observe that almost all the material dissolved after only 7 days of immersion, but there was a progressive increase of the pore wall thickness due to the deposition of HCA (observed also through SEM, see Fig. 5). All these findings assume that material dissolution in SBF caused density variations. In particular, a less dense area identified a portion of dissolved material and appeared darker than the non-dissolved zones.6 The HCA phase grew over time on the surface of the foam which was clearly visible as a whiter layer (because of its higher X-ray absorbance compared to dissolved BG).

µ-CT 2D reconstructions of BG-SA, BG-SL and BG-PU scaffolds before and after immersion in SBF for up to 28 days (a), pore size variation of BG-SA (b), and BG-SL (c) scaffolds after immersion in SBF for up to 28 days.
A broader distribution of pore sizes was observed for BG-SA and BG-SL scaffolds immersed in SBF for up to 28 days, as shown in Figs. 6(b) and 6(c). This was observed up to 3 weeks immersion in SBF. However, after this time-point the pore size distribution did not change further, most probably due to the combination of BG dissolution and HCA precipitation.
In addition, it was confirmed that the pore interconnectivity of the starting scaffolds did not change over time, both BG-SA and BG-SL scaffolds were characterized by a high interconnected pore structure (>99%).
Cell Culture Test
Indirect cell tests were conducted in order to evaluate potential cytotoxicity of the dissolution products of the BG-based scaffolds. The toxicity test showed that all the extraction volumes obtained from immersing the non pre-treated BG-based scaffolds for 1 day had a deleterious effect on cell viability if not diluted (i.e. 100%) (Fig. 8). However, the diluted conditioned medium (50%) which was in contact with the BG-SA samples showed high cell viability comparable with that of the control (no significant viability reduction, p < 0.05). Conditioned medium diluted at 10 and 1% showed the same cell viability compared to the control for all the samples.

(a) Cell viability results from indirect cell culture test with different dilutions of the extraction volumes, SEM micrographs of (b) BG-PU, (c) BG-SA and (d) BG-SL scaffolds after 14 days of direct cell culture study, (e) cell viability results from direct cell culture up to 14 days.
Saos-2 cells were also directly seeded onto BG-PU, BG-SA and BG-SL scaffolds in 24-well culture plates. Considering the cells adhered to the specimens, SEM observations revealed normal cell morphology after 14 days of culture with clear evidence of active cell attachment over the scaffold surfaces (Figs. 8b–8d). The cell viability on the BG-PU and BG-SL scaffolds was significantly lower when compared to the control, while the viability of cells seeded on the BG-SA samples was comparable to the control. For BG-SA scaffolds, no significant differences were found in cell viability values obtained on neither the inhalant nor the exhalant surfaces of the scaffolds.
Discussion
Currently, 45S5 BG, the first composition of bioactive glasses developed by Hench in the late 1960s,18 is still the most-used glass formulation for clinical applications as a bulk or powder material. Three-dimensional BG-based scaffolds are still not available for load-bearing applications due to their poor mechanical properties and lack of mechanical stability with high porosity (>90%).24 In fact, it is still a challenge to develop bioactive glass-based 3D porous scaffolds with high and open porosity and suitable mechanical properties, which should be as close as possible to those of natural bone.24 In a previous work, the possibility to improve the mechanical behaviour of BG-based scaffolds was considered using natural marine sponges, SA and SL, as sacrificial templates.7 The resulting scaffolds were characterized by increased compressive strength (up to 4 MPa), which was achieved due to a reduction in the total porosity (68–76%). This was achieved without affecting the pore interconnectivity (higher than 99%), compared to scaffolds prepared with PU foams as their template.9 Due to the lower porosity, the oxygen diffusivity of the BG-based scaffolds derived from natural marine sponges was lower when compared to that of the BG-PU scaffolds.5 It was found that the oxygen diffusivity of the natural marine sponges-based scaffolds increased over time due to the degradation of the material after 1 month in SBF.5 These structures are, in fact, expected to dissolve in the body, hence a time-dependent oxygen diffusivity can be anticipated.
In the present work, the bioactivity and the mechanical stability of these BG-based scaffolds derived from natural marine sponges were assessed and a comparison with the traditional BG-PU scaffolds was possible. As a consequence of the immersion of the BG scaffolds in SBF up to 28 days, a decrease in weight and an increase in total porosity of the BG-SA and BG-SL scaffolds was observed. At the end of the test, BG-SA and BG-SL scaffolds respectively lost 40 and 50% of their weight due to the degradation process of the bioactive glass. Interesting, almost the whole weight loss occurred during the first week of immersion in SBF, while in the subsequent 3 weeks the weight values remained almost constant. This behavior was more pronounced on the BG-SL samples and was most probably a consequence of the established equilibrium, being achieved after 7 days in SBF, due to the degradation of the BG and the precipitation of HCA on the surface of the scaffolds. In addition, the porosity changed continuously during the entire period of the test and a plateau was reached only between 3 and 4 weeks of immersion in SBF. One possible explanation for this apparent mismatching between weight and porosity variations can be the different densities of BG and HCA. The material deposited on the surface of the scaffolds is characterized by a higher density compared to that of the BG struts. It should be noted that throughout the SBF test, no variation in the geometry of the scaffolds was observed over time. During the first 3 weeks of immersion in SBF, both degradation and precipitation occurred but degradation was the dominant phenomenon. After 3 weeks, the two mechanisms reached equilibrium and the results indicate that at higher immersion times in SBF, precipitation could be the dominating mechanism until the HCA completely covered the structure of the scaffolds (Fig. 5). This behavior is in agreement with previous modeling work reported in the literature27 and similar results were recently obtained for Bioglass®-based scaffolds produced via powder metallurgy-inspired technology.6
As a consequence of the porosity increase, the compression strength of the BG-SA and BG-SL scaffolds decreased. After 28 days in SBF, the resulting compressive strength values were 1.2 ± 0.1 MPa for the BG-SA and 0.23 ± 0.01 MPa for the BG-SL scaffolds, these values were still higher than those of as-sintered BG-PU scaffolds (<0.05 MPa).7 In the present case, most of the compressive strength loss took place during the first week of immersion in SBF and afterwards the values were almost constant. This result is in agreement with the measured weight and porosity variations of the scaffolds. One possible explanation is that due to the higher density of the HCA deposit compared to the degraded BG struts, even if the total porosity was increased, a strengthening of the scaffolds was induced. Moreover, since the HCA formation commenced at the areas of the scaffolds with higher concentrations of cracks, these sites probably acted as favorable nucleation points, when exposed to the SBF solution. It is possible that the HCA crystals filled these cracks strengthening of the overall scaffold structure.
As shown by the ICP results (Fig. 4), the dissolution of the glass took place immediately after immersion in SBF inducing a rapid increase in the pH, mainly due to the fast exchange of Na+ and Ca2+ with H+ and H3O+ ions from the solution. This effect caused the hydrolysis of the silica groups and consequently, the formation of silanol groups which are known to be the starting point for the nucleation of HCA.13 Due to this very fast reactivity of BG, it was possible to identify the presence of a deposit on the surface of the scaffolds already after 1 day in SBF. This deposit evolved in thickness and after 14 days it was possible to identify the crystallization of the deposit (HCA), as confirmed by FTIR spectra. This deposit did not grow only on the external surface of the scaffolds but also on the surfaces of the inner core of the BG-foams, as confirmed by the micro-CT analysis. These observations highlight the high level of interconnectivity of the porous network of all produced samples. Another interesting aspect is that the pore size distribution became broader for both BG-SA and BG-SL scaffolds. Both smaller and larger pores increased in number, especially at 14 days of immersion in SBF. Moreover, even though HCA precipitation was significant, the smaller porosity was not occluded, suggesting homogeneous degradation of the scaffolds in the entire volume.
The high surface reactivity of non-pre-treated BG based scaffolds also resulted in cell culture test showing a low viability of cells cultured for 24 h in the non-diluted extraction medium. In the case of the BG-SA scaffolds, already after a dilution of 50%, no significant viability reduction was observed (p < 0.05) compared to the control. Moreover, the direct cell culture test on BG-SA samples showed that cells grew similarly on both surfaces of the scaffold (inhalant and exhalant) and the cell viability was comparable to that on the control. In the case of BG-PU and BG-SL scaffolds, the cell viability was significantly lower, most probably due to the higher pH values around the scaffolds. This result indicated that in these scaffolds the preconditioning treatment was not sufficient to avoid the formation of an undesirable non-physiological pH.
Conclusions
The bioactivity and mechanical stability of BG-based scaffolds derived from natural marine sponges was demonstrated. The obtained samples were characterized not only by improved mechanical properties (compressive strength up to 4 MPa) compared to the foams prepared using PU foam as a template, but also by the fact that after immersion in SBF the mechanical properties were stable for up to 7 days of testing. Moreover, the reduction of total porosity of the BG-SA and BG-SL scaffolds did not affect the bioactivity and already after 1 day of test in SBF, a deposit of HCA (the marker of bioactivity) was detected. Preliminary cell culture tests demonstrated that no toxic residues came from the natural marine sponges used as templates and cells proliferated extensively on the scaffolds. In conclusion, the best results in terms of mechanical properties, bioactivity and cell biocompatibility were obtained using “Spongia Agaricina” as template for BG scaffolds. These foams represent thus a new attractive family of BG-based scaffolds for bone tissue engineering warranting further in vitro and in vivo studies.
References
Aguilar-Reyes, E. A., C. A. León-Patiño, B. Jacinto-Diaz, and L. P. Lefebvre. Structural characterization and mechanical evaluation of bioactive glass 45S5 foams obtained by a powder technology approach. J. Am. Ceram. Soc. 95(12):3776–3780, 2012.
Amini, A. R., C. T. Laurencin, and S. P. Nukavarapu. Bone tissue engineering: recent advances and challenges. Crit. Rev. Biomed. Eng. 40(5):363–408, 2012.
Arkudas, A., A. Balzer, G. Buehrer, I. Arnold, A. Hoppe, R. Detsch, P. Newby, T. Fey, P. Greil, R. E. Horch, A. R. Boccaccini, and U. Kneser. Evaluation of angiogenesis of bioactive glass in the arteriovenous loop model. Tissue Eng. Part C 19(6):479–486, 2013.
Arrington, E. D., W. J. Smith, H. G. Chambers, A. L. Bucknell, and N. A. Davino. Complications of iliac crest bone graft harvesting. Clin. Orthop. Relat. Res. 329:300–309, 1996.
Boccardi, E., I. V. Belova, G. E. Murch, A. R. Boccaccini, and T. Fiedler. Oxygen diffusion in marine-derived tissue engineering scaffolds. J. Mater. Sci. Mater. Med. 26:200, 2015.
Boccardi, E., V. Melli, G. Catignoli, L. Altomare, M. T. Jahromi, M. Cerruti, L.-P. Lefebvre, and L. De Nardo. Study of the mechanical stability and bioactivity of Bioglass® based glass-ceramic scaffolds produced via powder metallurgy-inspired approach. Biomed. Mater. 11:015005, 2016.
Boccardi, E., A. Philippart, J. A. Juhasz-Bortuzzo, G. Novajra, C. Vitale-Brovarone, and A. R. Boccaccini. Characterisation of bioglass based foams developed via replication of natural marine sponges. Adv. Appl. Ceram. 114(S1):S56–S62, 2015.
Cerruti, M., D. Greenspan, and K. Powers. Effect of pH and ionic strength on the reactivity of Bioglass® 45S5. Biomaterials 26(14):1665–1674, 2005.
Chen, Q. Z., I. D. Thompson, and A. R. Boccaccini. 45S5 Bioglass®-derived glass-ceramic scaffolds for bone tissue engineering. Biomaterials 27(11):2414–2425, 2006.
Cunningham, E., N. Dunne, S. Clarke, S. Y. Choi, G. Walker, R. Wilcox, R. E. Unger, F. Buchnan, and C. J. Kirkpatrick. Comparative characterization of 3-D hydroxyapatite scaffolds developed via replication of synthetic polymer foams and natural marine sponges. J. Tissue Sci. Eng. S1:001, 2011. doi:10.4172/2157-7552.S1-001.
Gerhardt, L.-C., and A. R. Boccaccini. Bioactive glass and glass-ceramic scaffolds for bone tissue engineering. Materials 3(7):3867–3910, 2010.
Gorustovich, A. A., J. A. Roether, and A. R. Boccaccini. Effect of bioactive glasses on angiogenesis: a review of in vitro and in vivo evidences. Tissue Eng. Part B Rev. 16(2):199–207, 2010.
Hench, L. L. Bioceramics: from concept to clinic. J. Am. Ceram. Soc. 74(7):1487–1510, 1991.
Hench, L. L. The story of bioglass. J. Mater. Sci. Mater. Med. 17(11):967–978, 2006.
Hench, L. L. Opening paper 2015-some comments on Bioglass: four eras of discovery and development. Biomed. Glas. 1:1–11, 2015.
Hench, L. L., and J. Wilson. Surface-active biomaterials. Science 226(4675):630–636, 1984.
Hoppe, A., N. S. Güldal, and A. R. Boccaccini. A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics. Biomaterials 32(11):2757–2774, 2011.
Jones, J. R. Review of bioactive glass: from Hench to hybrids. Acta Biomater. 9(1):4457–4486, 2013.
Kokubo, T., and H. Takadama. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27(15):2907–2915, 2006.
Liu, W., T. Wang, Y. Shen, H. Pan, S. Peng, and W. W Lu. Strontium incorporated coralline hydroxyapatite for engineering bone. ISRN Biomater, 2013. http://dx.doi.org/10.5402/2013/649163.
Macon, A. L. B., T. B. Kim, E. M. Valliant, K. Goetschius, R. K. Brow, D. E. Day, A. Hoppe, A. R. Boccaccini, I. Y. Kim, C. Ohtsuki, T. Kokubo, A. Osaka, M. Vallet-Regí, D. Arcos, L. Fraile, A. J. Salinas, A. V. Teixeira, Y. Vueva, R. M. Almeida, M. Miola, C. Vitale-Brovarone, E. Verné, W. Höland, and J. R. Jones. A unified in vitro evaluation for apatite-forming ability of bioactive glasses and their variants. J. Mater. Sci. Mater. Med. 26:115, 2015.
Miguez Pacheco, V., L. L. Hench, and A. R. Boccaccini. Bioactive glasses beyond bone and teeth: emerging applications in contact with soft tissues. Acta Biomater. 13:1–15, 2015.
Peitl, O., E. D. Zanotto, and L. L. Hench. Highly bioactive P2O5-Na2O-CaO-SiO2 glass-ceramics. J. Non Cryst. Solids 292(1–3):115–126, 2001.
Philippart, A., A. R. Boccaccini, C. Fleck, D. W. Schubert, and J. A. Roether. Toughening and functionalization of bioactive ceramic and glass bone scaffolds by biopolymer coatings and infiltration: a review of the last 5 years. Expert Rev. Med. Devices. 12(1):93–111, 2015.
Porter, J. R., T. T. Ruckh, and K. C. Popat. Bone tissue engineering: a review in bone biomimetics and drug delivery strategies. Biotechnol. Prog. 25(6):1539–1560, 2009.
Pronzato, R., and R. Manconi. Mediterranean commercial sponges: over 5000 years of natural history and cultural heritage. Mar. Ecol. 29:146–166, 2008.
Sanz-Herrera, J. A., and A. R. Boccaccini. Modelling bioactivity and degradation of bioactive glass based tissue engineering scaffolds. Int. J. Solids Struct. 48(2):257–268, 2011.
Stevens, M. M. Biomaterials for bone tissue engineering. Mater. Today 11(5):18–25, 2008.
Vitale-Brovarone, C., E. Verné, L. Robiglio, P. Appendino, F. Bassi, G. Martinasso, G. Muzio, and R. Canuto. Development of glass-ceramic scaffolds for bone tissue engineering: characterisation, proliferation of human osteoblasts and nodule formation. Acta Biomater. 3(2):198–208, 2007.
Xynos, I. D., M. V. J. Hukkanen, J. J. Batten, L. D. Buttery, L. L. Hench, and J. M. Polak. Bioglass® 45S5 stimulates osteoblast turnover and enhances bone formation in vitro: implications and applications for bone tissue engineering. Calcif. Tissue Int. 67(4):321–329, 2000.
Acknowledgments
The authors would like to acknowledge financial support from the EU ITN FP-7 project “GlaCERCo”. The authors would like to acknowledge Dr. Judith A. Juhasz-Bortuzzo for correcting the English of this paper.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Akhilesh K. Gaharwar oversaw the review of this article.
Rights and permissions
About this article

Cite this article
Boccardi, E., Philippart, A., Melli, V. et al. Bioactivity and Mechanical Stability of 45S5 Bioactive Glass Scaffolds Based on Natural Marine Sponges. Ann Biomed Eng 44, 1881–1893 (2016). https://doi.org/10.1007/s10439-016-1595-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-016-1595-5