
Abstract
The invasion of malignant glioblastoma (GBM) cells into healthy brain is a primary cause of tumor recurrence and associated morbidity. Here, we describe a high-throughput method for quantitative measurement of GBM proliferation and invasion in three-dimensional (3D) culture. Optically clear hydrogels composed of thiolated hyaluronic acid and gelatin were chemically crosslinked with thiol-reactive poly(ethylene glycol) polymers to form an artificial 3D tumor microenvironment. Characterization of the viscoelasticity and aqueous stability indicated the hydrogels were mechanically tunable with stiffness ranging from 18 Pa to 18.2 kPa and were resistant to hydrolysis for at least 30 days. The proliferation, dissemination and subsequent invasion of U118 and U87R GBM spheroids cultured on the hydrogels were tracked in situ with repeated fluorescence confocal microscopy. Using custom automated image processing, cells were identified and quantified through 500 µm of gel over 14 days. Proliferative and invasive behaviors were observed to be contingent on cell type, gel stiffness, and hepatocyte growth factor availability. These measurements highlight the utility of this platform for performing quantitative, fluorescence imaging analysis of the behavior of malignant cells within an artificial, 3D tumor microenvironment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is a grade IV astrocytoma with particularly poor clinical prognosis. Following surgical resection, radiation treatment, and chemotherapy, patients face a median survival time of only 12–15 months.38 Invasion of malignant cells from the bulk tumor into surrounding healthy brain tissue remains a key factor in tumor recurrence and lethality, since the cells evade surgical resection and are also often resistant to chemotherapy and radiation therapy.17,34
In cancer research, the majority of standard migration assays are performed on two-dimensional (2D) tissue culture plastic. However, it is becoming increasingly understood that cells interact differently with three-dimensional (3D) substrates as compared to 2D surfaces.4,21,25 For example, in 3D, GBM cells interact with the extracellular matrix (ECM) through various membrane surface receptors, while also secreting enzymes, such as matrix metalloproteinases (MMPs). These enzymes actively degrade ECM glycoproteins, glycosaminoglycans (GAGs) and fibrous proteins to facilitate invasion.4 Moreover, the tumor–ECM interactions initiate complex cell fate-regulating signaling pathways that are drivers of invasion and tumor malignancy.3,11,24
Both natural and synthetic materials have been used to construct customizable microenvironments to study the movement of cells in 3D.4 Of the naturally derived materials, hyaluronic acid (HA) is of particular interest for studying cellular behaviors in GBM. HA is a negatively charged GAG polysaccharide of d-glucuronic acid and N-acetylglucosamine repeat units that is the most abundant component of brain ECM.6,18 The interaction of malignant cells with HA plays a significant role in regulating migration through the ECM, and is mediated by cell surface receptors such as CD44 and RHAMM.1,3,6,18,29 Expression of these receptors is frequently upregulated in high grade gliomas, and the concentration of HA is higher in the in vivo tumor microenvironment compared to healthy brain.1,11 These factors synergistically stimulate increased GBM–HA interactions to promote malignancy.3,18
Multiple approaches have been taken to develop HA-based scaffolds with tunable properties that incite biologically relevant invasion in vitro. 2,9,15,19,22,32,36,37,40 For example, Pedron et al. reported that glioma cells cultured in HA hydrogels lacking integrin binding domains will form spontaneous spheroids.32 An additional consideration in the design of HA scaffolds is that they are susceptible to degradation by enzymes that are secreted by cells and may weaken during extended 3D cultures. Consequently, most HA systems are combined with other materials that provide cellular adhesion sites, such as gelatin, or those that improve enzymatic stability, such as poly(ethylene glycol) (PEG). Interestingly, thiol-ene crosslinking with acrylate terminated PEG has been widely used to improve the enzymatic stability of HA hydrogels,22,26,33 even though the ester linkage may be susceptible to hydrolytic degradation.13 HA-composite scaffolds have been used to study cell morphology, gene expression, and protein expression of malignant cells over time.2,15,22,32,36 While this prior work provides compelling rationale for studying GBM behavior in 3D, few platforms are capable of quantitatively tracking both proliferation and invasion over time using a single assay format.
The goal of this work was to develop a better material platform and analytical method for studying GBM invasion in vitro. Hyaluronic acid and gelatin were functionalized with thiol groups to allow for chemical crosslinking with thiol-reactive PEG crosslinkers, including PEG diacrylate (PEGDA) and PEG divinyl sulfone (PEGDVS). Cell proliferation and invasion were tracked over time in situ with repeated fluorescence confocal microscopy and were quantified from this data using a custom, automated image analysis program. We observed distinct cellular behaviors that were a function of cell type, microenvironmental stiffness, and the availability of hepatocyte growth factor (HGF). Improved methods for tracking and measuring GBM invasion in vitro could provide insight into the cues that promote invasion, divergence from the proliferative tumor, and chemoresistance. Future work will focus on the development of this biomaterial platform to study the response of patient-derived cells to anti-invasive or chemotherapeutic agents, opening the door to personalized GBM therapy.
Materials and Methods
Materials
3,3-Dithiopropionic acid (DTPA), anhydrous methanol, anhydrous ethanol, hydrazine hydrate (HH), hexane, gelatin type A from porcine skin, hyaluronic acid sodium salt (HA) from Streptococcus equi, N-3-dimethylaminopropyl-N′-ethylcarbodiimide hydrochloride (EDC), polyethylene glycol (PEG) 6 kDa, dichloromethane (DCM), ethyl ether, triethylamine (TEA), acryloyl chloride, and 5,5′-dithiobis-2-nitrobenzoic acid (Ellman’s reagent) were purchased from Sigma Aldrich (St. Louis, MO, USA). Dithiothreitol (DTT) was purchased from Gold Biotechnology (St. Louis, MO, USA). Poly(ethylene glycol) divinyl sulfone (PEGDVS) 5 kDa was purchased from JenKem Technology USA (Allen, TX, USA). Dulbecco’s modified eagle medium (DMEM), fetal bovine serum (FBS), recombinant human hepatocyte growth factor (HGF), and trypsin–EDTA were purchased from Life Technologies (Carlsbad, CA, USA).
Polymer Synthesis
Dithiopropionic dihydrazide (DTPH) was synthesized from DTPA and HH in a two-step reaction as previously described.40 High molecular weight HA and gelatin were functionalized with thiol groups through conjugation of DTPH via EDC chemistry using a previously described procedure.37 PEGDA was synthesized from PEG through an acyl halide addition reaction with acryloyl chloride.13 1H NMR spectra were collected for each polymer product with D2O as the solvent for HA and gelatin and CDCl3 as the solvent for PEGDA (400 MHz Varian liquid state NMR, Agilent Technologies, Santa Clara, CA, USA). Ellman’s reagent test was used to confirm and quantify the thiolation of the HA and gelatin polymers.14 Dried polymers were sterilized by ethylene oxide gas and stored at −20 °C until used.
Preparation of HA-Gelatin Hydrogels
Hyaluronic acid (HA-S) and gelatin (Gel-S) hydrogels were chemically crosslinked via Michael-type addition with PEGDA or PEGDVS.13,30 Gel-S and HA-S were dissolved in equal concentrations in pH 3 cell culture media (DMEM + 10% FBS) and titrated between pH 7 and pH 8 with 1 M NaOH to deprotonate acid groups on the polymers, using phenol red as a colorimetric indicator of pH. PEG crosslinker (either PEGDA or PEGDVS) was dissolved in DMEM + 10% FBS at concentrations that yielded an equimolar ratio of thiol-reactive groups to thiols present on the HA-S and Gel-S. HA-S and Gel-S solutions were combined in a 1:1 volumetric ratio and mixed with an equal volume of PEG crosslinker solution. The combined solution was vortex mixed for 15 s prior to plating.
Rheometry
The mechanical properties of hydrogels with varying total polymer content were evaluated by rheometry. Immediately after mixing HA-S/Gel-S with the crosslinker, 400 µL of gelling solution was placed between the flat 25 mm plates of an Anton Paar MCR-101 rheometer (Anton Paar USA Inc, Ashland, VA, USA) preheated to 37 °C with a gap height of 0.5 mm. Hydrogels were formed at 5.00, 3.54, 2.50, 1.77, 1.25, 0.88, and 0.63 w/v% combined HA-S/Gel-S concentration. Gel concentrations were selected beginning with 5.00 w/v% and decreasing the w/v% by a factor of √2 until the concentration was too dilute to form a gel. A measurement frequency of 1 Hz was used with an oscillatory shear strain in the linear viscoelastic region (0.5%). The storage and loss modulus of each gel concentration corresponding to the viscoelastic stiffness and viscosity were recorded for up to 6 h after mixing. The “working time” for each hydrogel was calculated as the time to reach a storage modulus of 10 Pa, above which it could no longer be transferred by a micropipette.
Hydrogel Hydrolysis
The hydrolytic stability of gels crosslinked with either PEGDA or PEGDVS was evaluated in cell-free conditions. Crosslinker solutions were titrated between pH 5 and pH 10 in one-unit increments. HA-S/Gel-S hydrogels (2.5 w/v%, 1 mL volume) were cross-linked in microcentrifuge tubes and left overnight, after which an equal volume of cell-free media was added to the surface of each gel. Fresh media was exchanged every 2–3 days for 30 days. Hydrogel integrity was assessed by inversion of the microcentrifuge tube.
GBM Cell and Spheroid Culture
Green fluorescent (GFP) expressing human neuronal GBM cell lines were generously gifted by the Translational Genomics Institute (TGen, Phoenix, AZ, USA) and included U118, and U87R. U87R are an invasive population of U87-MG cells derived through serial in vivo passages of invasive subpopulations as previously described.23 Multicellular spheroids were formed in a hanging drop culture.10 Briefly, cells were suspended for 4 days in droplets of culture media (5 k cells/droplet) on the lid of a petri dish.
In Vitro 3D GBM Invasion Assays
Spheroids were cultured on the surface of hydrogels to assess cell invasion into the scaffold. Hydrogels were formed as described above at 2.5, and 1.77 w/v% HA-S/Gel-S and crosslinked with PEGDVS. Gels were formed in 70 μL volumes in a clear-bottom 96-well plate. The plates were incubated at 37 °C for 1 h to permit gelation of the scaffold prior to spheroid addition. A single GBM spheroid was gently pipetted onto the surface of each gel and allowed to attach overnight. An equal volume of media was added to each gel and exchanged every 2–3 days. For analysis of growth factor effect on invasion, HGF was added to the media at 20 ng/mL in selected samples. The hydrogel samples were incubated at 37 °C and 5% CO2, and were imaged with a Leica SPE II fluorescence confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with a live-cell incubator. 3D z-stack images were acquired at each time point starting at the top surface of the hydrogel and working down through 500 µm of gel in 10 µm steps to capture the spheroid and cells invading into the gel below (3D renderings of z-stack data at different time points are provided in Supplementary Videos 1-4). The initial position of each spheroid was marked in the imaging software at the first time point. At all subsequent time points, spheroids were located using the previously marked position, and imaged at that location. The growth of some spheroids at later time points extended beyond the microscope field of view. In these cases, the field of view was adjusted to capture the majority of the spheroid along with the cells invading into the gel below.
Image Processing
Images of GBM cells invading through the hydrogel were analyzed in MATLAB (2013B, MathWorks Inc., Natick, MA, USA) using native and scripted image processing functions to perform a watershed transformation to define individual cell borders.7 Example images showing a step-by-step representation of the program image outputs are provided in Supplementary Fig. 2. Total cell numbers were calculated in each image of the collected z-stacks as a function of depth in the hydrogel. A detailed description of the scripts developed to process images is provided in the Supplementary Information.
Statistics
Statistical tests were carried out in GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). Cell counts for all samples were compared between cell lines over time using 2-way ANOVA statistical testing, with repeated measures where applicable and Tukey post hoc tests in all cases. Statistical significance was evaluated with multiplicity adjusted p values and is reported where p < 0.05.
Results
Polymer Synthesis and Characterization
Synthesis of Gel-S and HA-S was confirmed by 1H NMR (Figs. 1a, 1b). The addition of thiols derived from DTPH side groups to HA and gelatin is indicated by the appearance of peaks at 2.5 and 2.7 ppm.37 Thiol content, determined by Ellman’s reagent test, was consistently measured in the range of 350–500 μmol per gram of polymer.14 PEGDA synthesis was also confirmed by 1H NMR (Fig. 1c), with acrylation of 84% to 100% of the available endgroups.13

1H NMR spectra of synthesized polymers. (a) Comparison of gelatin and thiolated Gel-S. The synthesis is confirmed by the appearance of two new peaks, highlighted in red, at 2.5 and 2.7 ppm. (b) Comparison of HA and thiolated HA-S. Thiolation is confirmed by the appearance of peaks, highlighted in red, at 2.56 and 2.75 ppm. (c) PEGDA (6 kDa) with acrylate-associated peaks, highlighted in red, appearing between 5.8 and 6.5 ppm, and the PEG-associated peak at 3.6 ppm
Hydrogel Preparation and Characterization
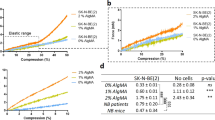
HA-S/Gel-S hydrogels were formed by chemical crosslinking available reactive thiols with PEGDA or PEGDVS, and the storage (G′) and loss (G′′) moduli for different formulations were determined by rheology (Table 1). G′ increased exponentially in the initial 30 min of gel formation, followed by slow crosslinking as remaining reactive groups were gradually consumed. G′′ was negligible in comparison to G′ in all formulations, indicating that the hydrogels are more accurately described by solid-like elastic behavior than by viscosity. PEGDA and PEGDVS crosslinked HA-S/Gel-S at the same concentration (2.5 w/v%) by comparable kinetics and reached similar resultant strength (Fig. 2a). Gels crosslinked with PEGDVS were also tested with HA-S/Gel-S concentrations ranging from 0.63 to 5.0 w/v%. As expected, lower total polymer concentration produced weaker gels, while the elastic strength increased with increasing concentration (Fig. 2b). At the highest concentration, 5.0 w/v%, gels crosslinked rapidly and the working time for hydrogel solutions was within 20 to 77 s (exact times within this range could not be determined due to lag in the initial rheometer measurement after plating the sample). These gels achieved a G′ of 18.2 kPa 6 h after mixing. Conversely, the lowest HA-S/Gel-S concentration yielding a measurable G′ was 0.88 w/v% at 18 Pa 6 h after mixing .

Viscoelastic characterization of HA-S/Gel-S/PEG hydrogels. (a) Comparison of the gelation profiles for 2.5 w/v% HA-S/Gel-S hydrogels crosslinked with either PEGDVS or PEGDA over 6 h. The gelation profiles are represented by the storage (G′) and loss (G′′) modulus values measured by rheology. (b) 30 min gelation profiles for hydrogels formed at concentrations of HA-S/Gel-S ranging from 0.88 to 5.0 wt.% crosslinked with PEGDVS (or PEGDA—denoted by **) represented by G′ values measured by rheology. As the concentration of polymers in the hydrogels increases, the speed of crosslinking and mechanical stiffness of the hydrogels increase
Hydrogel Hydrolysis
In early experiments, HA-S/Gel-S hydrogels crosslinked with acrylate terminated PEGDA were observed to dissolve upon incubation at 37 °C after several days, even in cell-free conditions. To test the hypothesis that dissolution of the scaffolds was due to hydrolysis of the acrylate-thiol linkage, we tested the integrity of 2.5 w/v% HA-S/Gel-S mixtures crosslinked either with PEGDA or with vinyl sulfone-terminated PEG (PEGDVS). The crosslinker solutions were titrated between pH 5 and 10 to analyze pH effects on gel stability. Hydrogels crosslinked with PEGDA in increasingly alkaline solutions degraded faster than gels formed in acidic solution. The most alkaline samples degraded within 9–12 days, and after 16 days, all PEGDA crosslinked gels had dissolved. Gels crosslinked with PEGDVS under the same conditions were observed for 30 days; these hydrogels did not dissolve, regardless of pH (Supplementary Fig. 1).
Cellular Invasion and Proliferation
GFP expressing multicellular spheroids (U118 or U87R, n = 6 for each group, 5000 cells per sample) were cultured on HA-S/Gel-S hydrogels crosslinked with PEGDVS for 14 days. Scaffolds were imaged regularly with fluorescence confocal microscopy to track spheroid growth and invasion of cells into the gel. GBM cells in each experimental sample were counted by automated analysis to determine cell number as a function of depth in the gel and time. The imaging setup allowed for cells to be counted in 50 discrete planar images that extended through 500 µm of the hydrogel. The image processing method compiled cell counts as a function of depth from the hydrogel surface. Representing average cell counts on the y-axis and relative depth from the surface of the gel on the x-axis, we were able to reconstruct profiles that characterized the proliferation and invasion of GBM cells through the hydrogel. A schematic of the experimental set up and measurement technique is displayed in Fig. 5a. Initial studies examined cell invasion on gels with stiffness varying from 18 Pa (0.88 w/v%) to 18.2 kPa (5.0 w/v%). Spheroids did not adhere well to 9.4 kPa or 18.2 kPa hydrogels, and appreciable growth was not observed in these constructs. The greatest distinction between proliferative and invasive behaviors over 14 days was observed in 2 kPa (1.77 w/v%) and 5 kPa (2.5 w/v%) gels, and these are the data presented here.
In 2 kPa hydrogels with standard media supplementation, U118 tumor spheroids were observed to rapidly proliferate and invade through more than 500 µm of gel within 10 days of culture (Figs. 3, 5b). Both rounded and elongated morphologies were observed, although the majority of invading cells were elongated. Increasing the gel stiffness to 5 kPa resulted in decreases in rates of proliferation and invasion as U118 spheroids grew as large solid structures with fewer detached invasive cells and fewer elongated cells (Fig. 3). The difference in growth is best identified by the altered shapes of the invasion curves, which flatten as cells invade deeper into the gel (Fig. 5c). Invasion in the stiffer 5 kPa gels was restricted to the upper ~ 300 µm of gel over 14 days. Supplementing cultures in 5 kPa hydrogels with HGF led to increases in invasion, as cells moved through more than 500 µm of gel by day 10 (Figs. 3, 5d). Thus, although increasing stiffness caused a decrease in invasion, a more invasive phenotype was adopted in the stiffer 5 kPa gels upon application of HGF.

Proliferative and invasive behavior of U118 multicellular GBM spheroids cultured in 3D HA-S/Gel-S/PEG hydrogels formed at 2, 5, and 5 kPa + HGF over 14 days. Fluorescence confocal images capturing GFP signal are shown as compressed 2D maximum intensity projections of a 3D image stack (step size = 10 µm, stack size = 500 µm, scale bar = 200 µm)
We next explored specific differences in proliferation relative to invasion. The average fold change in total cell number and maximum depth of invasion (500 µm) were measured as a function of time for each condition. Cells cultured in the 2 kPa gels showed significant increases in proliferation over the 5 kPa and 5 kPa + HGF cultures from days 8 through 14 (Fig. 6a). During that same time period, cells also invaded significantly deeper into 2 kPa and 5 kPa + HGF gels compared to non-supplemented 5 kPa gels (Fig. 6b). There were no differences in invasion depth between the 2 kPa and HGF supplemented 5 kPa hydrogel cultures at these time points.
The 2 kPa hydrogels supported rapid proliferation and invasion of U87R cells. In fact, U87R spheroids appeared to disseminate within 5 days of 3D culture such that the spheroid boundary was difficult to identify, with invading cells extending beyond the microscope field of view (Fig. 4). These cells invaded through more than 500 µm of gel within 8 days (Fig. 5e). Again, both rounded and elongated morphologies were observed, with most invasive cells exhibiting elongated spindle-shaped phenotypes. Increasing the gel stiffness to 5 kPa decreased the rates of proliferation and invasion (Fig. 4). Although cells at the rim of the spheroid were observed to move into the gel, this invasion was limited to the upper 250 µm of gel (Fig. 5f). Additionally, more invading cells appeared to adopt rounded morphologies compared to those observed in the softer gels. Similar to U118 cultures, addition of HGF produced an increase in the rate of invasion, with cells moving through ~ 450 µm of gel by day 14 (Fig. 5g).

Proliferative and invasive behavior of U87R multicellular GBM spheroids cultured in 3D HA-S/Gel-S/PEG hydrogels formed at 2, 5, and 5 kPa with supplemented HGF (20 ng/mL) over 14 days. Fluorescence confocal images capturing GFP signal are shown as compressed 2D maximum intensity projections of a 3D image stack (step size = 10 µm, stack size = 500 µm, scale bar = 200 µm)

Quantification of proliferative and invasive behaviors of U118 and U87R cells over 14 days in 3D culture. (a) Schematic detailing the experimental setup and resulting quantitative analysis. Multicellular spheroids and invasive cells were tracked by fluorescence imaging and counted through the depth of the hydrogels at each time point. Data is plotted as the average number of cells vs. distance from the gel surface (depth). (b + e) Average invasion profiles of U118 and U87R cells cultured in 2 kPa hydrogels. (c + f) Average invasion profiles of U118 and U87R cells cultured in 5 kPa hydrogels. (d + g) Average invasion profiles of U118 and U87R cells cultured in 5 kPa hydrogels supplemented with HGF (20 ng/mL)
Analysis of the invasion curves shows that proliferation was significantly increased in 2 kPa gels over 5 kPa gels from day 8 to day 12, and also in 5 kPa + HGF cultures at day 14 day (Fig. 6c). Similar to U118 cultures, invasion of U87R cells in 2 kPa gels was significantly greater than in 5 kPa gels from day 5 to day 14 (Fig. 6d). HGF also significantly increased the invasion of U87R in 5 kPa hydrogels at day 8 and also from day 12 to day 14. At the two later time points, there was no difference in invasion in 2 kPa and 5 kPa + HGF gels. Therefore, HGF enhanced both proliferation and invasion in the U87R cells in the 5 kPa hydrogel.

Analysis of U118 and U87R proliferation and invasion in 3D culture. (a + c) Proliferation is described by the fold change in average U118 and U87R cells counted in 500 µm stacks of confocal imaging data over 14 days. Data is normalized to the average total cells on day 1 and compared between the three microenvironmental conditions over time. (b + d) Invasion is described by the average maximum depth of cell penetration at each time point for the three microenvironmental conditions. (n = 6 for 5 kPa, n = 3 for 5 kPa + HGF and 2 kPa, *p < 0.05 compared to 5 kPa, $ p < 0.05 compared to 5 kPa + HGF, error bars = SEM)
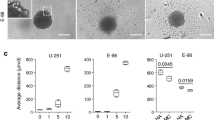
One interesting observation from these experiments was the formation of secondary spheroidal (multicellular) structures, which were observed for both U118 and U87R cells when spheroids were cultured on the surface of 5 kPa gels for 14 days (Fig. 7). These secondary structures were detached from the primary spheroid, and were not observed in either the 2 kPa samples or the 5 kPa samples supplemented with HGF. For the data in Fig. 7, we measured the shortest distance to the edge of the secondary spheroids from the edge of the primary spheroid in the same XY plane, as well as the largest diameter of each of these structures. In the pictured U118 sample, the primary spheroid has a long-axis diameter of 965 µm, and the four secondary spheroids have the following distance and diameter measurements from left to right as marked by the white arrows: 1. 526 µm distance, 144 µm diameter; 2. 465 µm distance, 154 µm diameter; 3. 651 µm distance, 137 µm diameter; 4. 259 µm distance, 188 µm diameter. For the U87R sample, the primary spheroid has a long-axis diameter of 619 µm, and the two secondary spheroids have the following measurements from top to bottom marked by white arrows: 1. 152 µm distance, 531 µm diameter; 2. 505 µm distance, 473 µm diameter.

Formation of secondary spheroids from invasive U118 and U87R cells within 5 kPa hydrogels after 14 days in culture. For both cell lines, the edge of the primary spheroid is circled with a white dotted line, and secondary spheroids are labeled with arrows. Images are compressed 2D maximum intensity projections of a 3D image stack capturing GFP signal (step size = 10 µm, stack size = 500 µm, scale bar = 200 µm)
Discussion
Unlike other cancers, glioblastoma does not metastasize through the blood stream and colonize other tissues.18,31 Instead, cells invade through the surrounding white matter and perivascular tissue to form lethal secondary tumors.31 These recurrent tumors often appear within several centimeters of the primary tumor location.28,34 Understanding the processes that modulate invasion and the response of invading cells to microenvironmental cues is therefore a principal issue in GBM research. Biomaterial scaffolds with tunable and biomimetic properties are useful tools for studying how malignant cells invade and interact with the local ECM. These systems can be engineered to recapitulate key aspects of the tumor microenvironment by providing a high degree of control over the physical and chemical properties of the matrix, such as adhesion site density, stiffness, and signaling factors.
In this work, we developed and characterized hydrolytically stable HA-S/Gel-S/PEG scaffolds suitable for in situ tracking of GBM proliferation and invasion in 3D with quantitative analysis of fluorescence imaging. HA is a highly relevant material for studying glioma invasion in vitro.2,22,32,36 Increased stromal HA content is a hallmark of the GBM microenvironment, and is directly correlated with increased tumor malignancy.11 The addition of proteins bearing enzymatically degradable focal adhesion sites, such as those found in gelatin, is necessary to elucidate invasive phenotypes in 3D cultures.2,32,33,36 Although fibrillar collagen is not a natural component of brain ECM, gelatin has previously been shown to promote physiological GBM behaviors in vitro.22,32,33 To study these malignant behaviors, we chose to culture two GBM cell lines, U118 and U87R, as models of noninvasive and invasive tumors respectively. The U118 cell line is a classic long-term immortalized GBM model originally isolated by Ponten and Macintyre.35 These cells have been described as forming solid tumors with well-defined boundaries in xenografts.8 The U87R cell line was derived from the parental U87-MG GBM cell line through isolation of tumorigenic cells that had invaded away from an orthotopic xenograft. This line was originally developed and characterized by Johnston et al. as a more biologically relevant model of GBM growth and invasion.23 Both cell lines were cultured as multicellular spheroids which are considered to more accurately represent native GBM structure, growth, and invasion compared to cell suspensions dispersed in 3D.10
Thiolation of HA and gelatin through EDC chemistry allows for participation in Michael-type addition reactions with a variety of Michael-acceptor reactive groups. These reactions are highly specific nucleophilic additions that occur under mild physiological conditions, without producing harmful byproducts. and can be initiated between thiols and terminal vinyl chemical groups such as acrylate, acrylamide, maleimide, and vinyl sulfone.13,30 Thiolated HA (HA-S) and gelatin (Gel-S) were chemically crosslinked with PEG functionalized with either acrylate (PEGDA) or vinyl sulfone (PEGDVS) end groups. This condensation polymerization strategy provided fast in situ gelation that is preferable to UV crosslinking commonly used to polymerize methacrylate-functionalized gelatin and HA.27,32,33 Michael-type addition does not require photoinitiator chemicals or generate free radicals and also allows for homogeneous crosslinking that is not limited by the penetration of light through the gel. The reaction is, however, nonreversible, and the hydrogel would require the addition hyaluronidase and gelatinase enzymes to be dissolved. The biochemical structure of the HA-S/Gel-S/PEG hydrogels offers improved reproducibility compared to natural ECM derivatives such as decellularized matrices and Matrigel®. Although the gel components do not recapitulate the complex composition or anisotropy of the native brain ECM, the resultant hydrogels formed optically clear 3D scaffolds that supported GBM spheroid growth, and enabled in situ measurements of proliferation and invasive cell penetration with confocal microscopy. This platform also offers the advantage of providing sites for incorporation of biologically relevant proteins, which will be an interesting subject for future studies.
The ideal scaffold format would maintain defined mechanical properties to facilitate analysis of cellular responses to mechanical cues. In early experiments, we observed significant hydrolysis in HA-S/Gel-S/PEG gels; scaffolds dissolved over several days, even in the absence of cells. Since local mechanical cues play an important role in determining the activity of malignant cells, we were motivated to develop a material platform for studying invasion that would both provide control over gel stiffness and maintain its integrity over the observation period. Both HA and gelatin are actively degraded by secreted matrix degrading enzymes, such as MMPs and hyaluronidase; these enzymes do not degrade the synthetic crosslinker poly(ethylene glycol) (PEG), which contributes to the popularity of PEG derivatives, such as PEG-diacrylate (PEGDA), for improving the structural support of scaffolds for 3D culture.22,26,33 However, we observed that PEGDA crosslinked HA-S/Gel-S hydrogels were susceptible to degradation even in the absence of cells (Supplementary Fig. 1). This degradation was caused by hydrolysis of the thio-acrylate linkages within the hydrogel which have also been empirically shown to degrade faster in alkaline solutions.13 The hydrolytic dissolution of PEGDA crosslinked gels confounds the interpretation of stiffness-dependent proliferation and invasion, and prevents analysis of these behaviors over time periods longer than several days. Therefore, PEGDA is an unsuitable crosslinker in this particular hydrogel system for studying the mechanobiology of GBM over long periods of time. By utilizing PEG-divinyl sulfone (PEGDVS) instead of PEGDA, we generated a stable scaffold capable of supporting the invasion of enzyme-secreting cells that was also resistant to hydrolysis. This stability allows for malignant cells to engage in behaviors, such as the formation of secondary spheroid structures, which may take up to 14 days to develop (Fig. 7).
Microenvironmental stiffness is recognized as a key controller of cellular phenotypes in 3D cultures and is directly related to polymer concentration.12 Gliomas cultured in 3D gels with lower stiffness and larger pore sizes will tend to exhibit increased motility with adherent cells characterized by their elongated morphologies and spine-shaped cellular processes.2,33,36 This phenotype is consistent with a mesenchymal mode of invasion in which cells enzymatically breakdown the local ECM.5,16 We further observed that cells adopted rounded morphologies in stiffer scaffolds. This behavior is consistent with an amoeboid mode of invasion in which cells rely on cytoskeletal rearrangement to squeeze through matrix pores, rather than degrading the ECM.5,16 Additionally, we observed that spheroids embedded within the 5 kPa hydrogels did not exhibit appreciable proliferation or invasion over the same time period (results not shown). The increased physical barrier of the matrix is a likely cause for this divergence. This has been previously reported in GBM cultures of similar stiffness, and investigated at length in other 3D in vitro cancer culture models.2,20,41 Polymer content—and resultant stiffness—is therefore an important microenvironmental cue that drives different mechanisms of motility. Rheological analysis of HA-S/Gel-S/PEG confirmed that these hydrogels behaved like solid elastic materials after crosslinking and that the stiffness of the gels increased with increasing concentrations of the constituent polymers and crosslinking density (Fig. 1). Matrix stiffness values from 18 Pa to 18 kPa were achieved by increasing or decreasing component concentrations (Table 1). Although the modulus of native brain is approximately 1 kPa, scaffolds similar to ours in stiffness (1–5 kPa) have been reported to induce biologically relevant proliferation and invasion in 3D GBM culture.2,15,32,33,36,39 Our results corroborate these reports.
Distinct qualitative differences in the extent and mode of invasion were observed between U118 and U87R in 3D culture (Figs. 3, 4). Spread morphologies, indicative of mesenchymal-type motility, were predominantly observed in both U87R and U118 cells cultured in 2 kPa hydrogels, in which cells rapidly invaded through more than 500 µm (Figs. 3, 4, 5b, and 5e). As the gel stiffness was increased to 5 kPa, both U118 and U87R cells exhibited decreased motility and proliferation. Additionally, rounded morphologies, suggesting amoeboid-type motility, predominated in the invasive populations. In 5 kPa cultures, U118 spheroids grew substantially, but did not invade into the deeper layers of the gel (Figs. 3, 5c). U87R cells were also not observed to invade deep into the 5 kPa gel, and generally moved radially across the upper gel layers where resistance to invasion was decreased (Figs. 4, 5f). The U87R line was developed by serial passaging of U87 cells in vivo to enrich an invasive subpopulation, and thus this result is not surprising. Moreover, U118 and U87R cells adopted a more invasive phenotype in 5 kPa gels when treated with exogenous HGF (Figs. 3, 4). HGF is a growth factor that promotes GBM proliferation and motility and is also commonly found at elevated concentrations in the tumor microenvironment.31 Supplementation of 5 kPa cultures with HGF increased U87R proliferation and increased invasion in both cell lines (Fig. 6). These observations confirm that the assay format developed here is capable of inducing and measuring biologically relevant cellular phenotypes.
After 14 days in 3D culture in the 5 kPa gels, invasive U118 and U87R cells began to form secondary clusters of cells within local proximity of the primary spheroid (Fig. 7). To our knowledge, this is the first report of the formation of secondary structures detached from a primary GBM spheroid maintained in 3D culture. These secondary structures could be composed either of cells that invaded as a group, cells that invaded to a similar location and subsequently adhered to one another, or individually invading cells that regained a proliferative phenotype. Secondary structures were only observed for spheroids maintained in culture for 14 days, highlighting the value of long term spheroid culture in 3D. Understanding the precise mechanisms by which secondary spheroids form in culture remains the subject of future work.
In this work, we describe the development of an in vitro platform for quantitative analysis of cellular invasion in a 3D microenvironment. HA-S/Gel-S hydrogels crosslinked with thiol reactive PEGDVS formed optically clear, hydrolytically stable scaffolds that supported cellular invasion for up to 2 weeks and the formation of secondary spheroids. The hydrogel platform enabled in situ quantitative measurements of cellular behaviors through fluorescence imaging and automated image processing. This technique could easily be expanded by reprograming the microscope to image larger volumes within each hydrogel, or by labeling protein or gene targets with fluorescent tags to quantify relative expression profiles. Such an expansion would further increase the applicability of this platform by enabling a broader range of biological analysis of cell–matrix interactions in a 3D paradigm. Cells were tracked with fluorescence confocal microscopy, and automated image analysis enabled in situ quantitative measurement of proliferation and invasion of cells through the depth of the gel. These methods were applied to identify distinct differences in invasive behavior as a function of cell type, matrix compositions, and growth factor availability. The platform developed here holds potential for future studies focused on the biological processes that define tumor cell malignancy and recurrence. Potential studies include but are not limited to the analysis of spatiotemporal aspects of secondary tumor spheroid formation, response of malignant cells to biophysical cues in the extracellular environment, or the identification of treatments that will most effectively target both invasion and proliferation in 3D.
References
Akiyama, Y., S. Jung, B. Salhia, S. Lee, S. Hubbard, M. Taylor, T. Mainprize, K. Akaishi, W. van Furth, and J. T. Rutka. Hyaluronate receptors mediating glioma cell migration and proliferation. J. Neurooncol. 53:115–127, 2001.
Ananthanarayanan, B., Y. Kim, and S. Kumar. Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials 32:7913–7923, 2011.
Ariza, A., D. López, J. L. Mate, M. Isamat, E. Musulen, M. Pujol, A. Ley, and J. Navas-palacios. Role of CD44 in the invasiveness of glioblastoma multiforme and the noninvasiveness of meningioma: an immunohistochemistry study. Hum. Pathol. 26:1144–1147, 1995.
Baker, B. M., and C. S. Chen. Deconstructing the third dimension–how 3D culture microenvironments alter cellular cues. J. Cell Sci. 125:3015–3024, 2012.
Beadle, C., M. C. Assanah, P. Monzo, R. Vallee, S. S. Rosenfeld, and P. Canoll. The role of myosin II in glioma invasion of the brain. Mol. Biol. Cell 19:3357–3368, 2008.
Bellail, A. C., S. B. Hunter, D. J. Brat, C. Tan, and E. G. Van Meir. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 36:1046–1069, 2004.
Beucher, S. The watershed transformation applied to image segmentation. Scanning Microsc.-Suppl. 6, 299–314, 1992.
Bullard, D. E., S. C. Schold, Jr, S. H. Bigner, and D. D. Bigner. Growth and chemotherapeutic response in athymic mice of tumors arising from human glioma-derived cell lines. J. Neuropathol. Exp. Neurol. 40:410–427, 1981.
Cheng, K., A. Blusztajn, D. Shen, T. S. Li, B. Sun, G. Galang, T. I. Zarembinski, G. D. Prestwich, E. Marbán, and R. R. Smith. Functional performance of human cardiosphere-derived cells delivered in an in situ polymerizable hyaluronan-gelatin hydrogel. Biomaterials 33:5317–5324, 2012.
Del Duca, D., T. Werbowetski, and R. F. Del Maestro. Spheroid preparation from hanging drops: characterization of a model of brain tumor invasion. J. Neurooncol. 67:295–303, 2004.
Delpech, B., C. Maingonnat, N. Girard, C. Chauzy, A. Olivier, R. Maunoury, J. Tayot, and P. Creissard. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumour stroma. Eur. J. Cancer 29:1012–1017, 1993.
Discher, D. E., P. Janmey, and Y. Wang. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 310:1139–1143, 2005.
Elbert, D. L., and J. A. Hubbell. Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules 2:430–441, 2001.
Ellman, G. L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70–77, 1959.
Florczyk, S. J., K. Wang, S. Jana, D. L. Wood, S. K. Sytsma, J. G. Sham, F. M. Kievit, and M. Zhang. Porous chitosan-hyaluronic acid scaffolds as a mimic of glioblastoma microenvironment ECM. Biomaterials 34:10143–10150, 2013.
Friedl, P., and K. Wolf. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3:362–374, 2003.
Giese, A., M. E. Berens, and M. Westphal. Cost of Migration: Invasion of Malignant Gliomas and Implications for Treatment. J. Clin. Oncol. 21:1624–1636, 2003.
Giese, A., and M. Westphal. Glioma invasion in the central nervous system. Neurosurgery 39:235–252, 1996.
Gurski, L. A., A. K. Jha, C. Zhang, X. Jia, and M. C. Farach-Carson. Hyaluronic acid-based hydrogels as 3D matrices for in vitro evaluation of chemotherapeutic drugs using poorly adherent prostate cancer cells. Biomaterials 30:6076–6085, 2009.
Guzman, A., M. J. Ziperstein, and L. J. Kaufman. The effect of fibrillar matrix architecture on tumor cell invasion of physically challenging environments. Biomaterials 35:6954–6963, 2014.
Hubbell, J. A. Biomaterials in Tissue Engineering. Nat. Biotech 13:565–576, 1995.
Jiglaire Jiguet. C., N. Baeza-Kallee, E. Denicolaï, D. Barets, P. Metellus, L. Padovani, O. Chinot, D. Figarella-Branger, and C. Fernandez. Ex vivo cultures of glioblastoma in three-dimensional hydrogel maintain the original tumor growth behavior and are suitable for preclinical drug and radiation sensitivity screening. Exp. Cell Res. 321:99–108, 2014.
Johnston, A. L., X. Lun, J. J. Rahn, A. Liacini, L. Wang, M. G. Hamilton, I. F. Parney, B. L. Hempstead, S. M. Robbins, and P. A. Forsyth. The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol. 5:e212, 2007.
Kenny, P. A., G. Y. Lee, C. A. Myers, R. M. Neve, J. R. Semeiks, P. T. Spellman, K. Lorenz, E. H. Lee, M. H. Barcellos-Hoff, and O. W. Petersen. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 1:84–96, 2007.
Kim, J. B., R. Stein, and M. J. O’Hare. Three-dimensional in vitro tissue culture models of breast cancer—a review. Breast Cancer Res. Treat. 85:281–291, 2004.
Lawyer, T., K. McIntosh, C. Clavijo, L. Potekhina, and B. K. Mann. Formulation Changes Affect Material Properties and Cell Behavior in HA-Based Hydrogels. Int. J. Cell Biol. 1–9:2012, 2012.
Leach, J. B., and C. E. Schmidt. Characterization of protein release from photocrosslinkable hyaluronic acid-polyethylene glycol hydrogel tissue engineering scaffolds. Biomaterials 26:125–135, 2005.
Lefranc, F., J. Brotchi, and R. Kiss. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J. Clin. Oncol. 23:2411–2422, 2005.
Merzak, A., S. Koocheckpour, and G. J. Pilkington. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 54:3988–3992, 1994.
Morpurgo, M., F. M. Veronese, D. Kachensky, and J. M. Harris. Preparation and characterization of poly (ethylene glycol) vinyl sulfone. Bioconjug. Chem. 7:363–368, 1996.
Nakada, M., S. Nakada, T. Demuth, N. L. Tran, D. B. Hoelzinger, and M. E. Berens. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 64:458–478, 2007.
Pedron, S., E. Becka, and B. A. C. Harley. Regulation of glioma cell phenotype in 3D matrices by hyaluronic acid. Biomaterials 34:7408–7417, 2013.
Pedron, S., and B. A. C. Harley. Impact of the biophysical features of a 3D gelatin microenvironment on glioblastoma malignancy. J. Biomed. Mater. Res. A 101:3404–3415, 2013.
Petrecca, K., M.-C. Guiot, V. Panet-Raymond, and L. Souhami. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J. Neurooncol. 111:19–23, 2012.
Ponten, J., and E. Macintyre. Long term culture of normal and neoplastic human glia. Acta Pathol. Microbiol. Scand. 74:465–486, 1968.
Rao, S. S., J. DeJesus, A. R. Short, J. J. Otero, A. Sarkar, and J. O. Winter. Glioblastoma Behaviors in Three-Dimensional Collagen-Hyaluronan Composite Hydrogels. ACS Appl. Mater. Interfaces 5:9276–9284, 2013.
Shu, X. Z., Y. Liu, F. Palumbo, and G. D. Prestwich. Disulfide-crosslinked hyaluronan-gelatin hydrogel films: a covalent mimic of the extracellular matrix for in vitro cell growth. Biomaterials 24:3825–3834, 2003.
Stupp, R., W. P. Mason, M. J. Van Den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, and U. Bogdahn. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352:987–996, 2005.
Ulrich, T. A., and E. M. de Juan. Pardo, and S. Kumar. The Mechanical Rigidity of the Extracellular Matrix Regulates the Structure, Motility, and Proliferation of Glioma Cells. Cancer Res. 69:4167–4174, 2009.
Vercruysse, K. P., D. M. Marecak, J. F. Marecek, and G. D. Prestwich. Synthesis and in vitro degradation of new polyvalent hydrazide cross-linked hydrogels of hyaluronic acid. Bioconjug. Chem. 8:686–694, 1997.
Yang, Y., S. Motte, and L. J. Kaufman. Pore size variable type I collagen gels and their interaction with glioma cells. Biomaterials 31:5678–5688, 2010.
Acknowledgments
1H NMR experiments were performed at Arizona State University at the Magnetic Resonance Research Center. The authors would like to acknowledge our colleagues Mike Berens, Nhan Tran, and Harshil Dhruv at the Translational Genomic Institute (TGen) in Phoenix, AZ for providing cell lines and advice, and Luis Laitano at Arizona State University for performing rheological analysis. The authors also acknowledge Arizona State University and Barrow Neurological Institute for providing funding support during this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Jennifer West oversaw the review of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Heffernan, J.M., Overstreet, D.J., Le, L.D. et al. Bioengineered Scaffolds for 3D Analysis of Glioblastoma Proliferation and Invasion. Ann Biomed Eng 43, 1965–1977 (2015). https://doi.org/10.1007/s10439-014-1223-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-014-1223-1