
Abstract
Biological invasions represent a major threat to agriculture and forestry across the globe. Thaumastocoris peregrinus is a small sap-sucking heteropteran bug that has recently invaded a number of eucalypt plantations worldwide from its native range in Australia. To date, no studies have examined the range of this insect within Australia, and its population genetics remain poorly understood. We sampled T. peregrinus from 16 populations from South East Queensland, across New South Wales (NSW) and Victoria to south-eastern South Australia, and generated microsatellite and mtDNA data for ~ 200 individuals. Population genetic analyses consistently revealed moderate levels of genetic isolation by distance among populations across the range. Nonetheless, T. peregrinus has undergone dispersal across large distances, as revealed by the presence of identical mitochondrial haplotypes in both South East Queensland and South Australia. Two populations within the Sydney area (NSW) were divergent from other populations based on STRUCTURE and factorial correspondence analysis. They also had relatively low allelic richness and haplotype diversity indices. These results suggest they are the result of a relatively recent invasion event, consistent with their discovery in 2001. Pairwise genetic distance analyses suggest that the source of the invasion may have been central NSW. Our study provides an important framework for understanding the biology of this pest in its native environment, and may have implications for determining how it has invaded multiple areas worldwide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Key Message
-
Thaumastocoris peregrinus is an emerging pest of eucalypt plantations worldwide; however, no studies have examined the population genetics of this species in its native range of Australia.
-
We sequenced both mitochondrial and microsatellite DNA from 16 T. peregrinus populations in order to investigate the level of genetic structure of this species across its native range, and test the hypothesis that individuals in Sydney represent a recent invasion.
-
We provide evidence that the insect in Sydney is distinct from other populations, and that it appears to be a recent invasion, whose source may be central NSW.
Introduction
Biological invasions represent a major threat to agriculture and forestry across the globe, particularly for crops and forest plantations grown outside their native range (Pimentel et al. 2001). Although commercially important plant species typically remain free of pests and disease for some time after being introduced to a foreign area, the organisms that attack them in their native range are often inadvertently introduced into the same areas (Hurley et al. 2016). Such exotic insect pests of crops and plantations often initially prosper due to a lack of natural enemies and competitors (Pimentel et al. 2001; Keane and Crawley 2002; Wingfield et al. 2008).
Eucalypts are the most widely planted hardwood tree species in the world (Rejmánek and Richardson 2011), with more than 20 million hectares established across Africa, Asia, Australia, Europe, and the Americas (Brockerhoff et al. 2006). They are prized for their rapid growth rate and adaptability to varying soils and climates (Hurley et al. 2016). Although early eucalypt forestry outside Australia benefitted from a lack of insect species that attack these trees (Wingfield et al. 2008), several pest insects endemic to Australia have now dispersed to areas where eucalypts are grown commercially (Paine et al. 2011; Hurley et al. 2016).
One such insect is Thaumastocoris peregrinus (Hemiptera: Thaumastocoridae), a small (2–3.5 mm) sap-sucking heteropteran bug from the subfamily Thaumastocorinae (family Thaumastocoridae). Three of the four genera of Thaumastocorinae are endemic to Australia, with the monotypic genus Wechina restricted to southern India. Of the Australian genera, Baclozygum and Onymocoris each contain four species while Thaumastocoris contains 14 species. Thaumastocoris has the broadest host range within the Thaumastocorinae, with eight plant families recorded as hosts; the majority of species are associated with Myrtaceae (Noack 2009; Cassis et al. 1999; Drake and Slater 1957).
Thaumastocoris peregrinus was described relatively recently, following outbreaks on planted Eucalyptus spp. trees in Sydney around 2001, South Africa in 2003, and Argentina in 2005 (Carpintero and Dellapé 2006). Since then, it has spread to Zimbabwe (2007), Brazil (2008), Malawi (2008), Uruguay (2008), Kenya (2009), Italy (2011), New Zealand (2012) (references in Saavedra et al. 2015), Israel (2015) (Novoselsky and Freidberg 2016), Mexico (2015) (Jiménez-Quiroz et al. 2016), and the USA (2016) (Hodel et al. 2016). It is thought to have invaded areas outside of Australia through infested plant materials, or through attachment on human clothing or other objects or commodities that are subject to long distance air travel and road transport (Wilcken et al. 2010; Nadel and Noack 2012).
Thaumastocoris peregrinus attacks at least thirty Eucalyptus species and three common commercial hybrids (Nadel et al. 2009). Laboratory studies established that, following a nymphal stage of 15–25 days, adults live a further ~ 16 days, during which females may lay up to 60 small black eggs usually on a single leaf (Noack and Rose 2007). Eggs hatch after 4–8 days, allowing nymphs and adults to occur on the same leaf (Noack and Rose 2007; Soliman et al. 2012). Following infestation, canopy leaves typically become reddened at first, and later develop a reddish-yellow or yellow brown colour (Noack 2002; Nadel et al. 2009). If infestation is severe, large-scale loss of leaves and canopy thinning may occur, in some cases leading to branch dieback and tree death. It appears that this damage is due to feeding by nymphs and adults on chloroplasts among other cell contents, leading to chlorosis (Santadino et al. 2017).
Systemic pesticides have been used to successfully control T. peregrinus in an urban setting; however, their use is not practical for large-scale plantations (Noack et al. 2009; Nadel and Noack 2012). Other potential control methods include insects that parasitize or feed on T. peregrinus (Mutitu et al. 2013). The parasitic wasp Cleruchoides noackae (Hymenoptera: Mymaridae) from Australia lays its eggs inside those of T. peregrinus (Noack 2002), and the neuropteran Chrysoperla externa (Neuroptera: Chrysopidae) was noted feeding on T. peregrinus nymphs in Brazil (Wilcken et al. 2010). The effectiveness of each of these candidate biocontrol agents has yet to be fully evaluated.
To date, there have been no extensive investigations of Australian populations of T. peregrinus. The relatively recent discovery of T. peregrinus on planted Eucalyptus species (namely E. nicholli and E. scoparia in 2001) in Sydney suggests that populations are not endemic to this area. The distribution of T. peregrinus across Australia is not completely understood. Nadel and Noack (2012) listed its distribution as occurring from South East Queensland, through much of New South Wales, and into Northern Victoria and the southern gulfs of South Australia (see Noack et al. 2011 for detailed collection locations).
In order to better understand the origins of invasive taxa, molecular markers can be generated from organisms in their native and invasive ranges. Nadel et al. (2009) sequenced a fragment of mitochondrial cytochrome oxidase I subunit from T. peregrinus from Sydney, south-eastern Queensland, South Africa, and Argentina. They found a total of eight haplotypes among Australian populations, three of which were found in invasive populations from outside of Australia. Their results indicated that there had been two independent invasions of T. peregrinus into South Africa and one into Argentina, each of which may have originated in Sydney. A better understanding of the distribution of T. peregrinus in Australia, as well as the population genetic structure of the species, could provide clues as to the origins of Sydney populations and provide a framework for understanding the origins of invasive populations outside Australia. Such information could also potentially provide information about where endemic biological control agents are present.
A series of microsatellite markers to examine the population genetics of T. peregrinus was developed by Gray et al. (2010). In this study, we collected T. peregrinus from a number of locations in central and southern New South Wales, Victoria, South Australia, and Queensland. We performed the first comprehensive study of the population genetic structure of T. peregrinus across its native range, using these microsatellite markers as well as the COI locus used by Nadel et al. (2009). An investigation of the origins of populations outside Australia using these same markers will be published in a future paper.
Materials and methods
Thaumastocoris peregrinus sampling, DNA extraction, and molecular marker analysis
Thaumastocoris peregrinus adults were sampled between 2004 and 2010 at 16 locations across Sydney, South East Queensland, central and southern NSW, Canberra, northern Victoria, and southern South Australia (Table 1). Based on the limited natural dispersal abilities expected of these insects, each collection location was considered a distinct population. Populations were further grouped into six regions (Table 1). Samples were taken from the reachable branches from a single tree at each location (in most cases on a street in a given town), identified using a field microscope, and placed in 100% ethanol. Specimens (all adults) were stored at either − 20 °C or − 80 °C until use. DNA was extracted from single individuals by homogenization with a sterile plastic pestle in a 1.5-ml Eppendorf tube, followed by boiling with 5% Chelex in 0.1 M TE buffer (Biorad).
Microsatellite genotyping and analyses
A total of 218 individuals were genotyped at seven polymorphic microsatellite loci following the methods outlined in Gray et al. (2010), using an ABI 3130xl Genetic Analyzer (Applied Biosystems). Tests for deviation of observed genotypes from Hardy–Weinberg Equilibrium for each microsatellite marker, and linkage disequilibrium between the markers, were carried out in FSTAT (Version 2.9.3.2; Goudet et al. 2002). The average number of different microsatellite alleles (Na), the observed (Ho) and unbiased (uHe) expected heterozygosities, and the fixation index (FIS) were calculated for each population using GenAlEx (Version 6.5; Peakall and Smouse 2012). Allelic Richness (AR; which uses a re-sampling procedure to eliminate biases that stem from uneven sample size) was also calculated using FSTAT for a standard sample size of eight individuals (the smallest number of samples with a complete microsatellite dataset at any single location, which was Coonabarabran). Null allele frequencies were estimated for each locus using the expectation maximization algorithm in the FreeNA package (Chapuis and Estoup 2007). Estimates of pairwise FST values with the ENA correction method of Chapuis and Estoup (2007) were calculated using FreeNA.
The structure of genetic variability in T. peregrinus was estimated using Wright’s F-statistics, calculated in GenAlEx. Genetic differentiation and relationships between the 16 populations were also estimated using pairwise FST (using both GenAlEx and FSTAT), Nei’s genetic distance (GenAlEx), and Factorial correspondence analysis (FCA; GENETIX). A Mantel test was carried out in GenAlEx to test for a correlation between genetic (linear genetic distances; LinGD) and geographic distance. FCA is a multivariate analysis method that is based on distance calculated between multi-locus genotypes of individuals, and does not make any genetic assumption regarding the populations studied. Lastly, STRUCTURE (Version 2.3.4; Pritchard et al. 2000) was used to assign individuals to predefined clusters by minimizing the Hardy–Weinberg disequilibrium and linkage disequilibrium within clusters. An admixture model with a 10,000 burn-in period and 100,000 MCMC repetitions was used. Different K values between K = 2 and K = 10, where K is the number of tested clusters, were applied, each for five iterations. The most likely number of clusters was determined using the method of Evanno et al. (2005), which is based on a number of criteria including: (1) the highest value of likelihood; (2) the trend of the likelihood distribution (plateau or decrease), and (3) the peak value of the ∆K.
Mitochondrial COI sequencing and analyses
A fragment of the mitochondrial COI gene (436 bp) from a total of 119 individuals was sequenced following the methods outlined in Nadel et al. (2009). Most of these individuals were also used for microsatellite marker analysis (see above), although in some cases (for example, where COI PCR or sequencing was unsuccessful) additional individuals were used. Sanger sequencing was performed by Macrogen (Seoul, Korea), and forward and reverse chromatograms were edited using Sequencher 4.5 (Gene Codes). No insertions, deletions, or stop codons were found among any of the obtained sequences. They could, therefore, be aligned unambiguously by eye. The COI sequences generated in this study were combined with 78 sequences from Sydney and Chelmsford from Nadel et al. (2009), to give a total of 197 sequences (Table 2), as well as appropriate outgroups (Thaumastocoris spp., see Nadel et al. 2009). Note that in Nadel et al. (2009) the seven samples labelled as ‘Brisbane’ in their Table 2 were actually from Chelmsford (~ 240 km northwest of Brisbane), which we re-sampled for this study (an additional 10 individuals, giving 17 in total in our Table 2). Nadel et al. (2009) combined the samples they collected across the Sydney area into the single category ‘Sydney’ (Table 2 of Nadel et al. 2009). For simplicity, we combined all of the COI haplotype data for their Sydney samples (from GenBank) with our data, and used the single category ‘Sydney’.
Phylogenetic analyses of a total of 197 ingroup and 6 outgroup (Thaumastocoris sp. 1 and sp. 2; Nadel et al. 2009) sequences were performed using maximum likelihood in MEGA6 (Tamura et al. 2013) and using Bayesian analysis in MrBayes 3.2.6 (Huelsenbeck and Ronquist 2001). The GTR + G model with six rate categories was used. Nearest neighbour interchange for the ML heuristic was selected, and 1000 bootstrap replications were performed. A total of 10,000,000 generations and two chains were run for Bayesian analysis, with 10,000 trees sampled and a burn-in of 1000 trees. Haplotype (h) and nucleotide (π) diversity were calculated for each population using DNASP (Librado and Rozas 2009). A haplotype network diagram was obtained using the software Popart (Leigh and Bryant 2015). A Mantel test was performed using Alleles in Space (Miller 2005), both including and excluding Sydney samples.
Results
Microsatellite data
A total of 218 individuals from 16 T. peregrinus populations were genotyped at seven microsatellite loci. The genetic diversity measures for each population are shown in Table 2. The mean number of alleles (Na) and allelic richness (AR) per locus across populations were 6.9 ± 0.4 and 6.0 ± 0.7, respectively. Mean observed (Ho) and unbiased expected (uHe) heterozygosity across populations were 0.56 ± 0.02 and 0.71 ± 0.02, respectively. The Sydney Sutherland population had the lowest values of genetic diversity (Na: 3.7 ± 0.5; AR: 3.4 ± 0.4; Ho: 0.4 ± 0.1; uHe: 0.6 ± 0.1), while Narrandera (NSW; Na: 11.1 ± 2.5) and Renmark (SA; AR: 6.6 ± 0.7; Ho: 0.7 ± 0.1; uHe: 0.8 ± 0.0) had the highest values. Despite being the most well-sampled population, Sydney Olympic Park (n = 40) had among the lowest values of AR, Ho, and uHe (Table 2).
Following the use of a Bonferroni correction for multiple comparisons, all populations were found to be in Hardy–Weinberg equilibrium for all loci except in 18 out of 112 tests. Most of these (10/18) were for locus Taus13. No pairs of loci were in significant linkage disequilibrium in more than two populations. For two loci, estimates for null allele frequencies were at or below 8% in at least 14 of 16 populations. A further three loci had estimated null allele frequencies at 8% or below in 10–12 of 16 populations. There were two loci in which estimated null alleles were greater than 8% in 11 and 15 of 16 populations, respectively.
Mean FIS (inbreeding coefficient) across populations was 0.2 ± 0.1 and ranged from − 0.02 ± 0.07 (Bacchus Marsh) to 0.30 ± 0.10 (Coonabarabran). The average FST value across all populations was 0.17 ± 0.03. Pairwise FST and Nei’s genetic distances are shown in Table 3 (see also Supplementary Table 1). There was a general trend for populations within the same region to have relatively low values for each of these measures, and for populations from geographically distant locations to have relatively high values. For example, pairwise FST values within the Central NSW region (Coonabarabran, Dubbo, Nyngan) ranged from 0.025 to 0.032, while values between Central NSW and SA ranged from 0.085 to 0.119. However, in a number of cases populations from distant locations had relatively low FST pairwise values (e.g. Narrandera and Renmark, 0.049), and some relatively proximate populations had higher FST pairwise values (e.g. Ararat and Bacchus Marsh; 1.24). Similar trends were found for Nei’s genetic distances (Table 3). The two Sydney populations had the lowest of all pairwise FST and Nei’s genetic distances (0.013 and 0.033, respectively). Sydney populations were most similar to samples from Coonabarabran (0.071–0.073 FST; 0.277–0.287 Nei’s distance) and other NSW populations (0.091–0.102 FST; 0.363–0.426 Nei’s). Estimates of pairwise FST values using the ENA correction method (Chapuis and Estoup 2007) were highly similar (within ~ 5–10%) of the uncorrected values shown in Table 3.
Factorial correspondence analysis of the microsatellite data is shown in Fig. 1. The Sydney populations formed a distinct cluster that was outside a larger cluster containing the other populations. Individuals from two populations, respectively, from Seymour and Wagga Wagga were found to be the greatest outliers within the larger cluster. The Sydney populations (Olympic Park and Sutherland) formed a separate cluster to the remaining populations. The results of Bayesian clustering conducted using STRUCTURE were congruent with the results from the FCA. A total of two clusters (one including the two Sydney populations and the other containing all other locations) were the most likely scenario according to Bayesian clustering in combination with the ∆K method (Fig. 2). In agreement with the pairwise FST and Nei’s genetic distance results, a relatively high proportion of individuals from Coonabarabran displayed multi-locus genotypes similar to those from Sydney (Fig. 2).

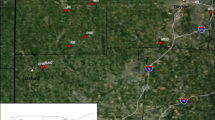
a Locations of each of the sixteen populations examined in this study (labelled 1–16 corresponding to locations in Table 1); b Factorial correspondence analysis (FCA) showing the clustering of T. peregrinus samples based on microsatellite data and according to region. Multi-locus scores are plotted in the bivariate space defined by the first two factorial components. Colours represent individuals from each region shown in Fig. 1a and Tables 1, 2, 3; the two Sydney populations (Olympic Park and Sutherland) have been labelled separately. Genetic relatedness is inversely proportional to the distance between points

Bayesian clustering of the multi-locus microsatellite genotypes of Thaumastocoris peregrinus individuals based on analysis in STRUCTURE. Each individual is denoted by a narrow vertical bar, and clustering is represented by colour (red or green). The number of clusters (K = 2; N = 218) was determined according to the Evanno et al. (2005) method. One hundred MCMC steps were performed, with a burn-in of 10,000 steps. Twenty iterations were performed. (Color figure online)
A Mantel test based on microsatellite data indicated a moderate level of genetic isolation by distance, both in analyses that included (r = 0.321; p < 0.001; data not shown) and excluded (r = 0.153; p < 0.001; Fig. 3a) samples from Sydney.

Mantel tests based on a microsatellite data and b mitochondrial COI data. In each case, Sydney samples were excluded from analyses, due to the likelihood that they are an invasive population. A significant correlation between geographic distance and genetic distance based on microsatellite data (linear genetic distances) was found (p < 0.001); however, this was not the case for mitochondrial genetic distance (p = 0.053)
Mitochondrial COI
Out of a total of 197 T. peregrinus COI sequences examined, 44 unique haplotypes were found. The proportions of the different haplotypes among the different populations are shown in Fig. 4, and a haplotype network is shown in Fig. 5. Sydney was the most extensively sampled region and had the highest number of haplotypes (9); however, several locations for which far fewer samples were obtained had multiple haplotypes [e.g. Cootamundra, NSW: five haplotypes; Renmark, SA: six haplotypes (Fig. 4)]. Haplotype D was the most common and widespread, being present in 10 of 15 populations sampled (Figs. 4, 5) (note Sydney Olympic Park and Sydney Sutherland were combined as one population). Haplotype A, which was separated from Haplotype D by only one base pair, was the second most widespread and common haplotype, being present in three populations (Figs. 4, 5). Haplotype diversity was lowest in Sydney (0.71) and reached 1.00 in Cootamundra, Narrandera, Seymour, Renmark, and Ararat; a value of 0.82 was found across all populations (Table 2). Nucleotide diversity ranged from 0.002 in Chelmsford up to 0.015 in Renmark, with a value of 0.0046 across all populations.

Proportions of mitochondrial haplotypes among Australian Thaumastocoris peregrinus across each population. Each haplotype with a shade or grey or pattern was found at more than one location. Each haplotype in white was found only at one site. The number of individuals examined at each site is given, and sites are grouped into regions

Mitochondrial haplotype network. Each mark represents one nucleotide difference. Circle sizes are in approximate proportion to haplotype frequencies. Letters inside or adjacent to each circle refer to haplotypes shown in Figs. 4 and 6. The distribution of each haplotype across populations is provided in Fig. 4. Haplotype D was the most commonly occurring haplotype (n = 54), followed by Haplotype A (n = 33), and Haplotypes G and H (n = 7 in each case). All other haplotypes were represented by either 1, 2, or 3 individuals (e.g. JB = 1, BC = 2, SC = 3)
A phylogenetic analysis of the 44 haplotypes revealed no obvious clustering of haplotypes by geographic region (Fig. 6). The earliest branching lineages contained populations collected at the western extreme of the T. peregrinus geographic distribution (LB from Renmark and L from Mitcham in South Australia), as well as Central Victoria (JB from Ararat) and NSW (W from Nyngan). The second earliest branching lineage contained representatives NSW and Victoria (B from Dubbo; BC, IB from Bacchus Marsh). The largest clade in Fig. 6 contained taxa from across the range of T. peregrinus.

Phylogeny of T. peregrinus from Australia based on mitochondrial COI sequences inferred using maximum likelihood. Each letter represents a different haplotype. Haplotypes A–H were identified by Nadel et al. (2009); all other haplotypes were identified in this study. Bootstrap values > 50% are shown at relevant nodes. The scale bar represents substitutions per site. Two closely related but uncharacterized Australian Thaumastocoris species (Nadel et al. (2009) were included as outgroups. A Bayesian analysis was also carried out in MrBayes 3.2.6. Posterior probabilities at those nodes supported by > 50% in ML analysis are shown below each value
A Mantel test comparing pairwise COI genetic distance with geographic distance indicated moderate levels of isolation by distance in analyses including Sydney samples (r = 0.31, p < 0.01), although less so in analyses that excluded Sydney samples (Fig. 3b; r = 0.095, p = 0.053).
Discussion
We found evidence for low to moderate levels of genetic differentiation among the sampled T. peregrinus populations, indicating that these insects do not have a strong natural ability to disperse (i.e. in the absence of human activity). Evidence for genetic isolation by distance was seen in pairwise FST and Nei’s genetic distance comparisons based on microsatellites, and also FST values for the population as a whole (0.16 ± 0.03), as well as in Mantel tests based on both microsatellite and mitochondrial data. (Although in the latter, the result was not significant in analyses excluding Sydney samples (p = 0.053).) The general trend for populations from neighbouring regions to have relatively low FST and Nei’s distances suggests that gene flow occurs more readily within regions (which encompass a diameter of ~ 250 km) than between regions (Hutchison and Templeton 1999).
The presence of representatives from one of the extremes of the T. peregrinus distribution (South Australia) in the earliest two branches in the mtDNA-based tree is consistent with some degree of genetic isolation by distance occurring in these insects. If there were higher levels of dispersal, the earliest branching lineages might be expected to come from a variety of areas throughout the distribution. As small insects of only a few millimetres in length, T. peregrinus might be expected to be actively fly only a few km (Zhang et al. 2008), although its flight may be assisted by wind. A relatively weak natural dispersal ability (i.e. without the aid of humans) might explain the moderate levels of genetic structure seen for T. peregrinus in our analyses.
Nonetheless, the distribution of identical mitochondrial haplotypes over wide geographic areas (e.g. Haplotype D from Coonabarabran and Ararat; Fig. 6) indicates that T. peregrinus has dispersed over distances of ~ 1000 km in the recent past. The mechanisms by which these insects have travelled these distances are unclear, but may be associated with natural phenomena such as strong winds (Drake and Farrow 1988; Chapman et al. 2010). That they are invasive insects in multiple countries shows that they also have the capacity to travel via human-related mechanisms (e.g. on seedlings/plants transported in trucks or cars moving between towns, in aircraft attached to human clothing, or on goods that are shipped to other countries).
Excess homozygosity (Ho) and slightly elevated levels of inbreeding (FIS) were found within a number of populations (e.g. Ararat, Coonabarabran, Narrandera, Renmark, Seymour, and Wagga Wagga). T. peregrinus has a generation time of ~ 40–50 days and is known to severely infest trees under some circumstances. If infestation is initiated by a limited number of individuals, a significant number of matings might occur between relatives. In this study, individuals were sampled in most cases from a single tree within a population, and thus had the potential to be close relatives, assuming that infestation leads to inbreeding. However, the mitochondrial data show that most populations have high mitochondrial diversity, and that individual maternal lineages do not dominate particular collection locations. This suggests that breeding between close relatives is not a common occurrence.
It is possible that a Wahlund effect (i.e. the presence of multiple, rather than single populations within each location examined) is responsible for excess homozygosity. However, given the expected short-range dispersal ability of this species (with the exception of human-mediated dispersal), it is unlikely that individuals present in the one stand of trees came from different populations, which could result in a Wahlund effect (Johnson and Black 1984). The biology of T. peregrinus, and particularly its breeding biology, is not well known. In aphids, populations in which facultative asexual reproduction occurs have markedly different inbreeding coefficients compared with those in which sexual reproduction occurs (Orantes et al. 2012). A better understanding of the breeding biology of T. peregrinus may provide insights into the levels of homozygosity and inbreeding in each population.
Perhaps the most likely explanation for the heterozygote deficiency we identified in a number of populations is the presence of null alleles (alleles that are not amplified due to mismatches in the primer regions). These were estimated to be at relatively high levels in two of the seven loci examined. Null alleles have been found to be associated with heterozygote deficiencies in a number of population genetics studies on a variety of organisms (Dabrowski et al. 2015). High numbers of null alleles have been found in a number of species, such as locusts, with large effective population sizes (Chapuis and Estoup 2007). Given the ability of T. peregrinus to rapidly infest trees, combined with its wide distribution, it might be expected this species has relatively large effective population sizes. Importantly, the potential presence of null alleles does not appear to having a large effect on our estimates of genetic distances between the different populations, based on corrections using the program FreeNA. The general concordance of results based on mitochondrial and microsatellite data also suggests that the latter are providing an accurate picture of the population of this species, despite the potential presence of null alleles.
Non-a priori population assignment using STRUCTURE and FCA was performed across all populations. The Sydney populations were found to have relatively low genetic diversity compared with the other populations despite the fact that nearly twice as many specimens were sampled compared with any other location (Table 2). The results from STRUCTURE and FCA suggest that these populations share unique characteristics compared with the others (Figs. 1, 2). This could be because the Sydney populations are the result of a recent invasion initiated by a limited number of individuals. Indeed, T. peregrinus was first observed in Sydney in 2001, and was hypothesized to have invaded within the last few decades (Noack 2002). If the Sydney population is invasive, one possible source of invasive individuals is a location in or near the Coonabarabran area. This population was found to be closely related to the Sydney population based on both FST and Nei’s genetic distances, and both populations contained the A, D, and M mitochondrial haplotypes.
The presence of pest organisms in areas of frequent travel and trade, such as Sydney, has been shown to significantly increase the chances of invasion in other regions (Hulme 2009; Estoup and Guillemaud 2010). In 2004, a few years following its discovery in Sydney, outbreaks were reported in various areas of South Africa and South America, and it has since spread to a number of other countries across the African, European, and North and South American continents. Future studies employing the use of combined microsatellite and mtDNA markers as well as approximate Bayesian computation (Csilléry et al. 2010) should provide an understanding of the possible invasion routes of T. peregrinus into these areas.
Author Contributions
NL, AN, and SL conceived and designed research. AM, AN, HW, and KR conducted experiments. RJN, HAR, ME, and GC contributed new reagents or analytical tools. AM, NL, HW, and HN analyzed data. NL wrote the manuscript. All authors read and approved the manuscript.
References
Brockerhoff EG, Bain J, Kimberley M, Knížek M (2006) Interception frequency of exotic bark and ambrosia beetles (Coleoptera: Scolytinae) and relationship with establishment in New Zealand and worldwide. Can J For Res 36:289–298
Carpintero DL, Dellapé PM (2006) A new species of Thaumastocoris Kirkaldy from Argentina (Heteroptera: Thaumastocoridae: Thaumastocorinae). Zootaxa 1228:61–68
Cassis G, Schuh RT, Brailovsky H (1999) A review of Onymocoris (Heteroptera: Thaumastocoridae), with a new species, and notes on hosts and distributions of other thaumastocorid species. Acta Soc Zool Bohem 63:19–36
Chapman JW, Nesbit RL, Burgin LE, Reynolds DR, Smith AD, Middleton DR, Hill JK (2010) Flight orientation behaviors promote optimal migration trajectories in high-flying insects. Science 327:682–685
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Csilléry K, Blum MGB, Gaggiotti OE, François O (2010) Approximate Bayesian Computation (ABC) in practice. Trends Ecol Evol 25:410–418
Dabrowski M, Bornelöv S, Kruczyk M, Baltzer N, Komorowski J (2015) ‘True’ null allele detection in microsatellite loci: a comparison of methods, assessment of difficulties and survey of possible improvements. Mol Ecol Res 15:477–488
Drake VA, Farrow RA (1988) The influence of atmospheric structure and motions on insect migration. Ann Rev Entomol 33:183–210
Drake CJ, Slater JA (1957) The phylogeny and systematics of the family Thaumastocoridae (Hemiptera: Heteroptera). Ann Entomol Soc Am 50:353–370
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19:4113–4130
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Goudet J, Perrin N, Waser P (2002) Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol 11:1103–1114
Gray KA, Noack AE, Johnson RN, Eldridge MDB, Bess EC, Lo N, Rose HA (2010) Eleven di-nucleotide polymorphic microsatellite markers for the winter bronzing bug Thaumastocoris peregrinus Carpintero and Dellapé (Heteroptera: Thaumastocoridae) Mol. Ecol Res 10:1106–1108
Hodel DR, Arakelian G, Ohara LM (2016) The Bronze Bug: another new threat to eucalypts in California. Palm Arbor 5:1–11
Huelsenbeck JP, Ronquist F (2001) MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hulme PE (2009) Trade, transport and trouble: managing invasive species pathways in an era of globalization. J Appl Ecol 46:10–18
Hurley BP, Garnas J, Wingfield MJ, Branco M, Richardson DM, Slippers B (2016) Increasing numbers and intercontinental spread of invasive insects on eucalypts. Biol Invasions 18:921–933
Hutchison DW, Templeton AR (1999) Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53:1898–1914
Jiménez-Quiroz E, Vanegas-Rico JM, Morales-Martínez O, Lomeli-Flores JR, Rodríguez-Leyva E (2016) First Record of the Bronze Bug, Thaumastocoris peregrinus Carpintero and Dellapé 2006 (Hemiptera: Thaumastocoridae), in Mexico. J Agric Urban Entomol 32:35–39
Johnson MS, Black R (1984) The Wahlund effect and the geographical scale of variation in the intertidal limpet Siphonaria sp. Mar Biol 79:295–302
Keane RM, Crawley MJ (2002) Exotic plant invasions and the enemy release hypothesis. Trends Ecol Evol 17:164–170
Leigh JW, Bryant D (2015) popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Librado P, Rozas Z (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Miller MP (2005) Alleles In Space (AIS): computer software for the joint analysis of interindividual spatial and genetic information. J Hered 96:722–724
Mutitu EK, Garnas JR, Hurley BP, Wingfield MJ, Harney M, Bush SJ, Slippers B (2013) Biology and rearing of Cleruchoides noackae (Hymenoptera: Mymaridae), an egg parasitoid for the biological control of Thaumastocoris peregrinus (Hemiptera: Thaumastocoridae). J Econ Entomol 106:1979–1985
Nadel RL, Noack AE (2012) Current understanding of the biology of Thaumastocoris peregrinus in the quest for a management strategy. Int J Pest Manag 58:257–266
Nadel RL, Slippers B, Scholes MC, Lawson SA, Noack AE, Wilcken CF, Bouvet JP, Wingfield MJ (2009) DNA bar-coding reveals source and patterns of Thaumastocoris peregrinus invasions in South Africa and South America. Biol Invasions 12:1067–1077
Noack AE (2002) Thaumastocoridae: an investigation. http://pandora.nla.gov.au/pan/32881/20030204/www.thaumastocoridae.org/index.html. Accessed 8 July 2011
Noack AE (2009) The taxonomic revision of the genus Thaumastocoris and the biology and chemical control of the eucalypt pest Thaumastocoris peregrinus (Heteroptera: Thaumastocoridae). Ph.D. thesis, The University of Sydney
Noack AE, Rose HA (2007) Life-history of Thaumastocoris peregrinus and Thaumastocoris sp. in the laboratory with some observations on behaviour. Gen Appl Entomol 36:27–33
Noack AE, Kaapro J, Bartimote-Aufflick K, Mansfield S, Rose HR (2009) Efficacy of imidacloprid in the control of Thaumastocoris peregrinus on Eucalyptus scoparia in Sydney, Australia. Arboricult Urb For 35:192–196
Noack AE, Cassis G, Rose HA (2011) Systematic revision of Thaumastocoris Kirkaldy (Hemiptera: Heteroptera: Thaumastocoridae). Zootaxa 3121:1–60
Novoselsky T, Freidberg A (2016) First record of Thaumastocoris peregrinus (Hemiptera: Thaumastocoridae) in the Middle East, with biological notes on its relations with eucalyptus trees. Isr J Entomol 46:43–55
Orantes LC, Zhang W, Mian MAR, Michel AP (2012) Maintaining genetic diversity and population panmixia through dispersal and not gene flow in a holocyclic heteroecious aphid species. Heredity 109:127–134
Paine TD, Steinbauer MJ, Lawson SA (2011) Native and exotic pests of Eucalyptus: a worldwide perspective. Annu Rev Entomol 56:181–201
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pimentel D, McNair S, Janecka J, Wightman J, Simmonds C, O’Connell C, Wong E, Russel L, Zern J, Aquino T, Tsomondo T (2001) Economic and environmental threats of alien plant, animal, and microbe invasions. Agric Ecosyst Environ 84:1–20
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rejmánek M, Richardson DM (2011) Eucalypts. In: Simberloff D, Rejmánek M (eds) Encyclopedia of biological invasions. University of California Press, Los Angeles, pp 203–209
Saavedra MC, Avila GA, Withers TM, Holwell GI (2015) The potential global distribution of the Bronze bug Thaumastocoris peregrinus Carpintero and Dellapé (Hemiptera: Thaumastocoridae). Agric For Entomol 17:375–388
Santadino M, Brentassi ME, Fanello DD, Coviella C (2017) First evidence of Thaumastocoris peregrinus (Heteroptera: Thaumastocoridae) feeding from mesophyll of eucalyptus leaves. Environ Entomol 46:251–257
Soliman EP, Wilcken CF, Pereira JM, Dias TKR, Zaché B, Dal Pogetto MHFA, Barbosa LR (2012) Biology of Thaumastocoris peregrinus in different eucalyptus species and hybrids. Phytoparasitica 40:223–230
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis, version 6.0. Mol Biol Evol 30:2725–2729
Wilcken CF, Soliman EP, Nogueira de Sá LA, Barbosa LR, Dias TKR, Ferreira-Filho PJ, Oliveir RJR (2010) Bronze bug Thaumastocoris peregrinus Carpintero and Dellapé (Hemiptera: Thaumastocoridae) on eucalyptus in Brazil and its distribution. J Plant Protect Res 50:201–205
Wingfield MJ, Slippers B, Hurley BP, Coutinho TA, Wingfield BD, Roux J (2008) Eucalypt pests and diseases: growing threats to plantation productivity. South For 70:139–144
Zhang Y, Wang L, Wu K, Wyckhuys KA, Heimpel GE (2008) Flight performance of the soybean aphid, Aphis glycines (Hemiptera: Aphididae) under different temperature and humidity regimens. Environ Entomol 37:301–306
Acknowledgements
The authors wish to thank Michael Brown for his generous financial assistance, which allowed the collections to take place, and Cleo Gardiner for helping with the collections.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by B. Hurley.
Special Issue: “Invasive Pests of Forests and Urban Trees”.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lo, N., Montagu, A., Noack, A. et al. Population genetics of the Australian eucalypt pest Thaumastocoris peregrinus: evidence for a recent invasion of Sydney. J Pest Sci 92, 201–212 (2019). https://doi.org/10.1007/s10340-018-0995-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-018-0995-8