
Abstract
The primary objective of this study is to implicate the Quality by Design (QbD) principle to develop a simple and stability—indicating High Performance liquid Chromatography (HPLC) method for analysing and quantifying Eltrombopag olamine. Initially, a comprehensive risk assessment was conducted using an Ishikawa (fish-bone) diagram. Following this, an Analytical Target Profile (ATP) was established, with desired specification and Critical Analytical Attributes (CAAs) were identified to fulfil these requirements. Additionally, Critical Material Attributes (CMAs) and Critical Process Attributes (CPPs) were chosen, as they can influence the CAAs. Subsequently, a three-level factorial design was utilized to optimize the primary contributing factors both numerically and graphically. The validation study was performed according to International Council for Harmonisation (ICH) guidelines and forced degradation studies were performed under various stress conditions. Optimal chromatographic separation was done using a mobile phase comprising acetonitrile and water with 0.3% formic acid in both phases at a ratio of 80:20% v/v, with 1.2 mL/min flow rate and UV detection at 248 nm. The developed method exhibited high sensitivity and specificity, with a linear range of 10–70 µg/mL and a correlation coefficient (R2) of 0.9999. It demonstrated accuracy with % recovery ranging from 98–100% and the detection and quantification limits of 0.2443 µg/mL and 0.7403 µg/mL, respectively. The forced degradation studies indicated that the drug is vulnerable to all stress conditions. Overall, the developed method proves to be suitable for estimation of Eltrombopag olamine in its marketed formulation, with potential applicability for analysing it in other dosage form and various biological samples available.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
QbD provides a structures approach to develop an analytical method prioritizing predetermined objectives to ensure the method reliability, robustness, and consistent delivery of accurate result [1]. This process involves defining the objective of analytical process, specifying the analysis purpose, target the analyte and desired performance outcome. A thorough risk assessment is conducted to identify critical method attribute and potential sources of variation that could affect the method performance [2, 3]. By employing systematic experimental design such as Design of Experiment the impact of various parameter on analytical method can be assessed and controlled. This enables the optimization of fundamental variables and critical method performance [4, 5]. ICH Q8 guideline is important and a foundational for QbD methodology in pharmaceutical development. It ensures quality of Analytical QbD for analytical method development through systematic approach [6, 7]. The stability indicating method (SIM) is important in pharmaceutical and quality control, because it provides an analytical tool for detecting and quantifying degradants or contaminants that may emerge during product shelf life or manufacturing process. Additionally, SIM provides all necessary information to meet the criteria of ICH regarding product stability [8]. This method also facilitates formulation development by elucidating degradation pathway, helps in selecting appropriate components and packaging materials, and provides information on decisions on reformulation or modification of storage conditions to maintain product quality and extend shelf life by monitoring stability characteristics [9, 10].
Eltrombopag Olamine (ELO) is a synthetic small, chemical molecule with a molecular formula C25H22N4O4.2(C2H7NO). The chemical name of the molecules is 3'-{(2Z)-2-[1-(3,4-Dimethylphenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene]hydrazino}-2'-hydroxy-3-biphenylcarboxylic acid-2-aminoethanol (1:2) [11]. To improve the solubility and stability, it is formulated as a salt form, Eltrombopag Olamine, as shown in (Fig. 1). It is used to treat low platelet count in patients suffering with conditions like chronic immune thrombocytopenia (ITP) and severe aplastic anaemia [12]. It acts as an orally active thrombopoietin receptor agonist that binds to c-Mpl (myeloproliferative leukemia protein) on megakaryocytes in the bone marrow [13]. It also activates JAK–STAT intracellular signaling pathway promoting differentiation and proliferation of megakaryocyte in the bone marrow region. Ultimately, this leads to significant increase in circulating platelets level [14].

Chemical structure of eltrombopag olamine
According to the literature review, there have been limited publications on HPLC-based method development for eltrombopag olamine. In one of the studies, the HPLC method is developed for the ELO in the presence of impurity and degradation [15]. In another study, the method was developed with QbD and performed degradation study on tablet dosage form [16]. These studies utilized buffer with various combinations and gradient flows to separate ELO which make the method complex. However, in another work on stability indicating HPLC methods for ELO estimation was done using UPLC but a QbD-based approach was not applied for method development, thus limiting their applicability [17]. All the research work showed variation in their degradation study. In the current work, a QbD enabled method is developed using an isocratic mobile phase which makes it simple and degradation study is performed on the pure drug substance. Isocratic elution, characterized by a constant mobile phase composition throughout the chromatographic analysis, offers simplicity and reproducibility, making it preferable choice for routine analysis. The degradation studies underscored the necessity of monitoring drug stability during formulation and storage, emphasizing the role of analytical methods in pharmaceutical development.
Experiment
Materials
ELO was provided as a gift sample by Glenmark Pharmaceuticals Limited. HPLC grade acetonitrile was purchased from Loba Chemie Pvt Ltd., India. Eltrog 25 mg tablets were purchased at the medical supply store. All other chemicals and reagents used were of analytical grade.
Equipments
The HPLC system used for method development and force degradation was Waters Alliance e2695 Separations Module. It is a system with a quaternary, low-pressure mixing pump and inline vacuum degassing. The detector is a photodiode array (model 2996) with a wavelength range of 190–800 nm. It processed and monitored the output signal using an Empower program. The Kromasil 100- 5C18 (250 × 4.6 mm) E80935 column was used for analysis.
The milli Q® system (Millipore, Milford, India) was used to obtain HPLC water, which was then further filtered using an Ultipor 0.45-µM membrane filter.
Standard Solution Preparation
The ELO pure drug was weighed accurately about 10 mg and transferred into a volumetric flask of 10 mL. Initially, it was dissolved in about a small quantity of ACN: Water (80:20) by vigorous shaking. Later, the same solvent was used to make a volume to mark 10 mL to prepare a standard stock solution of 1000 µg/mL. To make a working standard solution of 100 μg/mL, 1 mL of the standard stock solution was diluted with 10 mL of the solvent.
Sample Solution Preparation
Twenty Eltrog 25 (Eltrombopag olamine tablets) were individually weighed and placed in a clean mortar for trituration. The powder equivalent of 25 mg of drugs was weighed and placed in a 50-ml volumetric flask. To obtain a stock solution of 500 µg/mL, the volume was made up with ACN: Water (80:20) solvent. The working sample solution was prepared by transferring 1 mL of the stock solution to a 10-mL volumetric flask. Using the same solvent, the volume was made up to 10 ml to prepare a working sample solution with concentration of 50 µg/mL.
Analytical Target Profile (ATP) and Critical Analytical Attributes (CAAs)
The Analytical Target Profile (ATP) serves as a blueprint in defining and outlining the specific quality characteristic required for the method development, similar to Quality Product Profile (QTPP) for the drug product. The purpose of defining the ATP is to ensure quality, safety, and application of method [18]. Additionally, it also covers details related to analytical method attributes, such as instrument specification, target drug, sample preparation requirements, and attributes regarding the analytical method itself. It outlines the measurement requirement for quality attribute to ensure confidence in the quality decision made from the result [19].
Critical Analytical Attributes (CAA) are the key parameters within the chromatogram that must be meet predefined criteria to ensure the method quality and suitability. These are measurable attributes that have direct impact on performance and reliability of method. These CAAs are crucial in defining the fitness of analytical procedure throughout its lifecycle [20]. For HPLC method, development example of CAAs includes retention time (Rt), number of theoretical plates (NTPs), tailing factors (Tf), peak purity, and resolution between two peaks, peak area and assay. By defining the ATP and identifying CAAs, developers can develop HPLC method to meet specific quality criteria and ensure robustness in analytical performance [21].
Risk Assessment and Screening Trial
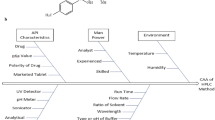
A risk assessment study was conducted to identify potential risk or failures in QbD-based method development. Using Ishikawa fish-bone diagram (Fig. 2) a cause-and-effect link between CMA, CPP and CAA was determined. This model aims to identify and select CMAs and CPPs that had a significant impact on CAAs [22]. For this study CMAs like the organic solvent ratio, percentage of acid while CPP like mobile phase, flow rate was determined. A standard solution of 10 µg/mL was selected for evaluation of the desired CAAs. For screening trial runs, three key variables like organic ratio of mobile phase (A), Formic acid concentration (B), and Flow rate of mobile phase (C) were evaluated. The organic ratio ranged from 60 to 80%, formic aid ratio varied between 0.1 and 0.3%, and flow rate was ranged from 0.8 to 1.2 mL/min. These parameters were carefully controlled within their selected range to ensure reliable and stable experimental condition. The key response variables were retention time (R1), area (R2), and number of theoretical plates (R3). The model suggested 27 runs with combinations of variables for screening studies.
Optimization Runs
Following the screening studies, formic acid concentration and flow rate were chosen for further optimization of analytical methods. The formic acid ratio varied between 0.1 and 0.3% and flow rate ranged from 0.8 to 1.2 mL/min. Optimization study was conducted using three-level factorial design, involving nine runs using a sample with 10 μg/mL standard concentration. The data obtained were analysed against key critical analytical attributes (NTPs and Rt). Statistical analyses from factor screening informed the method optimization conducted using Design Expert Version 13. The model efficiency was evaluated based on parameters such as correlation coefficient (R2, adjusted R2, predicted R2) and adequate precision.
Method Validation
The method validation parameters were performed and evaluated as per the current ICH guideline.
Specificity: Specificity assesses the ability of an analytical method to measure the accurate analyte of interest in presence of other components like excipients in the formulation, impurities, degradation product. It is determined by comparing standard solution of ELO and sample solution of marketed tablets of ELO at 50 µg/mL concentration.
Linearity: Linearity determines the relationship between the sample concentration and analytical response over a defined range of concentration. It is determined by plotting a calibration curve of a series of varying concentrations made from standard solution. A serial dilution from the standard solution was prepared in a concentration range of 10–70 µg/mL to evaluate the linearity. A graph of peak area against concentration was plotted to get the calibration curve. For every concentration, triplicate readings were taken.
Precision: Precision measures the degree of repeatability or reproducibility of the test result. The parameter of repeatability was determined using six readings at a concentration range of 10 µg/mL. Intraday precision was evaluated for 0, 2, and 4 h at different concentrations of 24, 30, and 36 µg/mL in triplicate, while interday precision was also evaluated for the same concentration. Precision studies were expressed in terms of percentage relative standard deviation (% RSD). This parameter ensures that the analytical method is consistent and reliable.
Accuracy: Accuracy refers to the closeness of test result values to the true values. The accuracy was determined by standard addition method. The standard solution of 30 µg/mL was prepared from a stock solution and 80%, 100%, and 120% of standard solution were spiked in the sample. The % recovery was calculated to determine accuracy.
LOD and LOQ: LOD is defined as lowest concentration of sample that can be reliably detected, while LOQ is defined as lowest concentration of sample that can be quantified. These parameters are determined by standard deviation of the response. LOD and LOQ were calculated using the following equations:
where σ is the standard deviation and S is the slope of the calibration curve of ELO.
Robustness: This parameter is used to evaluate the method capacity to remain unaffected by small variations like temperature, pH, and mobile phase composition in method parameter. These deliberate changes in method will help to assess the flexibility and reliability under different conditions. The robustness was studied at 10 µg/mL concentration by changing parameters like column temperature (± 5 °C, flow rate (± 0.3 mL/min), and mobile phase composition (± 5%).
Forced Degradation Study
Forced degradation studies in HPLC involve subjecting a compound to various stress conditions like acid, base, oxidation, photolysis, and thermal stress to identify degradation products. This also helps to understand the intrinsic property of the compound and to identify the degradation pathway and degradation products. Standard samples were prepared at 100 µg/mL and subjected to various stress conditions. A 10 µg/mL was prepared as a control sample for analysis. Before injection, the samples were neutralized for analysis if required.
Acid Degradation Studies
The solutions were prepared using 0.1 M, 1 M and 2 M HCL and stored in room temperature protected from light. The samples were withdrawn at different intervals of time and diluted as per the mentioned procedure.
Base Degradation Studies
The solutions were prepared using 0.1 M, 1 M and 2 M NaOH for basic degradation studies and stored in room temperature protected from light. The samples were withdrawn at different intervals and diluted as per specified procedure.
Oxidative Degradation Studies
The solutions were prepared using 3% H202 and stored in room temperature protected from light. The sample was withdrawn and diluted as per specified procedure.
Thermal Degradation Studies
The sample was exposed to a temperature of about 80 °C. The sample was withdrawn after 4 h and diluted to final concentration as procedure mentioned above.
Photodegradation Studies
The sample was placed in a UV light chamber and exposed to UV radiation. Afterward, sample was taken out over a 15 h period and diluted as procedure mentioned above.

Ishikawa (fish-bone) diagram
Results and Discussion
Risk Analysis and Screening Trial
To comprehend the correlation between CMAs and CPP and their impact on CAA, conducting risk assessment is crucial. The potential risk factors have been summarized in the Ishikawa fish-bone diagram. For factor screening studies, a few factors have been identified and selected for the study derived from risk assessment. The primary aim of this screening study was to identify the key factors that have significant influence on method development with minimal experimentation. After screening trials, among all the factors, two factors, specifically organic phase composition and flow rate, have significant impact on the responses, such as retention time, peak area, and NTFs Additionally, the interaction of organic phase composition and flow rate also has impact on responses, as shown in Fig. 3. As flow rate and organic ratio increases the retention time decreases. As flow rate decreases the area increases and as organic ratio increases the area increases. While increasing flow rate and organic ratio, the number of theoretical plates decreases. The formic acid composition does not have a significant impact on all responses. The actual equations for responses of screening runs are listed down in Table 1, while the analysis report summary is mentioned in Table 2.


a–c Screening Run 3D surface graphs of responses retention time (a), peak area (b), and number of theoretical plates (c)
Method Optimization Runs
Based on screening runs, the factors, such as formic acid concentration and flow rate, were considered for optimization study to get the optimum condition for method development. In this study, the factors were analysed against the response of retention time and NTPs shown in Fig. 4 as a 3D surface graph. For the analysis, the constraints were selected to get the desired result. The formic acid concentration was set to maximum to get a symmetric factor (tailing factor) in the limit range. The flow rate was set to maximum to get the earliest retention of peak. The retention time was set to minimum to reduce organic consumption.

a–b Optimization Run 3D surface graphs of responses retention time (a) and number of theoretical plates (b)
In this study, Three-Level Factorial Design was utilized for optimizing the chosen independent variables. Design Expert software recommended a linear model as the most suitable fitting model for two responses: Number of Theoretical Plates (NTPs) and Retention Time (Rt). The actual equations for responses are mentioned in Table 3. In Table 4, the R2 value of 0.9953 and the adjusted R2 value of 0.9922 suggest an excellent fit of the model to the data, indicating that the polynomial equation accurately represents the relationship between the independent variables and the retention time. Additionally, the predicted R2 value of 0.9839 and the Adequate Precision value of 31.2377 indicate that the model can effectively predict retention time under various conditions. However, for the Count variable, the coefficient of variation is higher at 2.36%, suggesting slightly more variability in the measurements compared to retention time. The R2 value of 0.8815 and the adjusted R2 value of 0.8025 indicate a good fit of the model to the data, though not as strong as for retention time. The lower predicted R2 value of 0.5752 suggests that the model may not be as reliable in predicting counts compared to retention time. In conclusion, the data indicate that the model developed for retention time is highly accurate and precise, while the model for count, although reasonably good, may have some limitations in its predictive capability. On the basis of the above constraint, the software provided four solutions. Among which the solution with flow rate of 1.2 mL/min and formic acid conc of 0.3% was giving the highest desirability of 0.968. The highest desirable solution was considered for method development. The chromatographic separation was confirmed by performing a trial as per desirable solution which matches with the outcome of the optimization study. This demonstrated the accuracy of the anticipated QbD model for the method development and achieving estimated CAAs.
The optimum chromatographic separation was achieved with mobile phase composition of ACN and water in 80:20 ratio (% v/v) with 0.3% formic acid with flow rate of 1.2 mL/min, column temperature at 25 °C and UV detection at 248 nm. This optimized method was validated as per the ICH guidelines.

a–b Chromatogram of ELO without excipient (a). Chromatogram of ELO with excipient (b)
Method Validation
Specificity
Specificity or sensitivity can be confirmed as the peaks are not interfering with each other; therefore, there is no significant effect of excipient on analysis of drug. As no peaks were found at the retention time of ELO. Specificity studies determined that there is no interference of excipient with drug peak where Fig. 5(a) shows chromatogram without excipient and Fig. 5(b) shows chromatogram with excipients.

Calibration curve of ELO
Linearity
A calibration curve was generated between the peak area (AU) and the analyte concentration (µg/mL) to verify the linearity of the developed analytical method. The correlation coefficient (R2) was 0.9999, indicating a strong relationship between observed and projected data as shown in Fig. 6.
Precision The acceptance criteria of %RSD states possessing a value below 2%, and the method shows good repeatability as the value of %RSD is 1.08%. The mean RSD (%) for Intraday was found to be 0.76, 0.29 and 1.03% and for interday, were found to be 0.203, 0.875, and 0.196% for 24, 30, and 36 μg/ml, respectively, as shown in Table 5. These findings highlighted the developed analytical method is highly precise.
Accuracy
The accuracy study was performed using standard addition method by adding 80%, 100%, and 120% of 30 μg/ml solution of ELO which resulted in 98–100% recovery, as shown in Table 6.
LOD and LOQ
The LOD and LOQ values were found to be 0.2443 µg/mL and 0.7405 µg/mL, respectively, for the developed analytical method. Thus, the developed method is highly sensitive, allowing for effective detection and quantification.
Robustness
Variations in several chromatographic parameters, including column temperature, mobile phase composition, and flow rate, resulted in negligible alterations in the CAAs, including NTP, Rt, and Tf. The insensitivity of responses to deliberate small changes in method conditions indicated the resilience of the developed HPLC method.
Analysis of Dosage Form
Label claim: Each pharmaceutical dosage form contains: 25 mg Eltrombopag Olamine.
The determination of ELO in commercially available formulations was established using % drug content computation after the development and validation of the method was done using HPLC. The recovery data for estimating ELO in pharmaceutical dosage form are presented in Table 7, and corresponding chromatogram is shown in Fig. 7.

Chromatogram of ELO tablet assay
Forced Degradation Study
In acidic conditions, initial treatment was done with 0.1 M, 1 M HCl which showed no remarkable degradation, whereas treatment with 2 M HCl for 2 h resulted in 29% degradation. In this condition, one degradant product was observed namely DP1 at Rt 3.43 min. In basic condition, one degradant product was observed at Rt 3.46 min. This degradant was noticed in 2 M NaOH for 2 h into 55% degradation. The drug was more susceptible in oxidative condition resulting in three degradant products, namely, DP2, DP3, and DP4 which were observed at Rt 1.62, 2.83, and 3.31 min, respectively. The degradants were observed under 3% H2O2 for 2 which resulted in 23%. Under thermal conditions, the DP5 was observed after exposure of 80 ºC for 4 h which resulted in 35% degradation of the drug. The degradant was noticed at Rt 3.5 min. In photolytic stress conditions, three degradant products, namely, DP6, DP7, and DP8, were observed at Rt 1.30, 1.41, and 2.72 min, respectively. Under photolytic conditions, the drug was degraded by 75%. The summary of degradation study is shown in Table 8, and chromatograms of different stress conditioned are shown in Fig. 8(a–e).


a–e Chromatogram of acidic condition (a), chromatogram of basic condition (b), chromatogram of oxidative condition (c), chromatogram of thermal condition (d), and chromatogram of photolytic condition (e)
Conclusion
A speedy and reliable QbD-based HPLC method to detect and measure ELO in both its pure form and different formulations. Initially, started by outlining the key quality requirements for developing the HPLC method, known as the Analytical Target Profile (ATP). Based on this, specific factors like retention time, peak area, and number of theoretical plates that are crucial were identified for method performance. Using Ishikawa fish-bone diagrams, different factors like mobile phase composition and process conditions like flow rate were figured out for the study. Then, fine-tuned these factors using a level factorial design to optimize the method. By drawing contour plots and surface plots, interaction between different factors with each other and with the key performance factors was understood. Afterward, numerical optimization was used to pinpoint the best analytical conditions, which was marked out with suggested desirability solutions. After that, thorough validation study including various parameters like linearity, sensitivity, accuracy, and robustness was carried out. The ELO was also subjected to different stress conditions to see how it degraded and found nine degradation products. Overall, the method proved its effectiveness and can be adapted for use with the other ELO formulations, showing the value of systematic approach. Furthermore, continuous improvement and optimization of method parameters could enhance sensitivity and shorten analysis times, thereby meeting the evolving needs of pharmaceutical analysis.
Data availability
No datasets were generated or analysed during the current study.
References
Mishra V, Thakur S, Patil A, Shukla A (2018) Quality by design (QbD) approaches in current pharmaceutical set-up. Expert Opin Drug Deliv. https://doi.org/10.1080/17425247.2018.1504768
Patel KY, Dedania ZR, Dedania RR, Patel U (2021) QbD approach to HPLC method development and validation of ceftriaxone sodium. Futur J Pharm Sci. https://doi.org/10.1186/S43094-021-00286-4
Susmitha A, Rajitha G, Eri GK (2023) A comprehensive review on QbD driven analytical procedures developed for the analysis of various drugs. J Liq Chromatogr Relat Technol 46(1–5):12–36
Katekar V, Sangule D, Bhurbhure O, Ingle P, Dhage S, Jadhav K (2022) A review on quality by design approach in analytical methods. J Drug Deliv Ther 12(3):255–261
Chaturvedi PK, Joshi KH, Chopra S, Tauro SJ, Prajapati PB (2023) Pitfalls and opportunities in the execution of quality by design in analytical sciences. Curr Pharm Anal 19(5):399–412
Bhavna BS, Ojha A (2022) International Council for Harmonisation (ICH) guidelines. Regulatory Affairs in the Pharmaceutical Industry. Elsevier, Amsterdam, pp 47–74
Attala K, Eissa MS, El-Henawee MM, Abd El-Hay SS (2021) Application of quality by design approach for HPTLC simultaneous determination of amlodipine and celecoxib in presence of process-related impurity. Microchem J 1(162):105857
Yadav A, Raghvendra D (2022) Stability indicating analytical method development and validation. Int J Pharm Sci Med 7(11):33–50
Sonawane JK, Chavan SM, Narkar IP, Jale SC, Tendulkar NV, ProfV J et al (2023) A review of stability indicating methods and forced degradation studies. Int J Res Publ Rev 4(5):4703–4715
Hsh M, Khalil Na A, Shehab A (2022) Stability of pharmaceutical preparations. MINAR Int J App Sci Technol 04(02):232–235
Corman SL, Mohammad RA (2010) Eltrombopag: a novel oral thrombopoietin receptor agonist. Ann Pharmacother 44(6):1072–1079
Fattizzo B, Levati G, Cassin R, Barcellini W (2019) Eltrombopag in immune thrombocytopenia, aplastic anemia, and myelodysplastic syndrome: from megakaryopoiesis to immunomodulation. Drugs 79(12):1305–1319
Bussel J, Kulasekararaj A, Cooper N, Verma A, Steidl U, Semple JW et al (2019) Mechanisms and therapeutic prospects of thrombopoietin receptor agonists. Semin Hematol 56(4):262–278
Cheng G (2012) Eltrombopag, a thrombopoietin- receptor agonist in the treatment of adult chronic immune thrombocytopenia: a review of the efficacy and safety profile. Ther Adv Hematol [Internet]. 3(3):155–164
Dandabattina R, Subramanian VB, Merugu KS, SharathBabu H, Katari NK, Kowtharapu LP (2023) Stability-indicating RP-HPLC method development and validation for eltrombopag olamine in the presence of impurities and degradation products. Robust Design Expert Softw J AOAC Int 106(2):267–275
Jayagopal B, Murugesh S (2021) QBD-driven HPLC method of eltrombopag olamine: degradation pathway proposal, structure elucidation, and in silico toxicity prediction. J Pharm Biomed Anal 203:114231
Patel AI, Gosai S, Jadav K, Vyas AJ, Patel AB, Patel NK (2020) Stability indicating analytical method development and validation of eltrombopag olamine in tablet dosage form by RP-UPLC. J Turkish Chem Soc Sect A: Chem 7(3):845–850
Verch T, Campa C, Chéry CC, Frenkel R, Graul T, Jaya N et al (2022) Analytical quality by design, life cycle management, and method control. AAPS J 24(1):34
Jackson P, Borman P, Campa C, Chatfield M, Godfrey M, Hamilton P et al (2019) Using the analytical target profile to drive the analytical method lifecycle. Anal Chem 91(4):2577–2585
Mishra V, Thakur S, Patil A, Shukla A (2018) Quality by design (QbD) approaches in current pharmaceutical set-up. Expert Opin Drug Deliv 15(8):737–758
Chavan AV, Gandhimathi R (2023) Quality by design approach: progress in pharmaceutical method development and validation. Biomed Pharmacol J 16(3):1669–1679
Deidda R, Orlandini S, Hubert P, Hubert C (2018) Risk-based approach for method development in pharmaceutical quality control context: a critical review. J Pharm Biomed Anal. https://doi.org/10.1016/j.jpba.2018.07.050
Acknowledgements
The authors are grateful to SVKM’s NMIMS SPPSPTM, Mumbai for providing the support and facilities to conduct this work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Godani, N., Mudaliar, S., Pai, R. et al. Leveraging Principles of QbD for Analytical Method Development and Validation for the Estimation of Eltrombopag Olamine in Tablet Dosage Forms by HPLC. Chromatographia (2024). https://doi.org/10.1007/s10337-024-04356-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10337-024-04356-6