
Abstract
To analyze 1-methyladenosine (m1A) and 1-methylinosine (m1I) in urine samples obtained from patients with breast cancer and healthy people, we used magnetic dispersive solid-phase micro-extraction (MDSPME) combined with high-performance liquid chromatography. The structure and morphology of magnetic nanoparticles, Fe3O4@GO, were investigated by FT-IR, XRD, TEM, and TGA. m1A and m1I in urine samples were enriched by the solid phase micro-extraction technology of Fe3O4@GO. Several parameters affecting the extraction efficiency, including adsorption time, pH, ionic strength, mass of adsorbent, extraction time, eluent solvent, elution time, and elution temperature, were researched and optimized. Under the optimized conditions, the corresponding linearity range of the two analytes showed a good linearity (r2 > 0.9998) and significant addition recoveries ranging from 88.50 to 109.38%, while the limit of detection was between 0.10 and 0.11 ng/mL. The enrichment factors of m1A and m1I were 108 and 134, respectively. The method was simple, sensitive, and reliable for the detection and purification with good detection limit, and it was suitable for the detection of m1A and m1I in urine samples. In addition, the analysis of modified nucleosides is of great value for the early diagnosis of patients with breast cancer.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Breast cancer is a common malignant tumor in women. It is internationally recognized that the “early prevention, early detection and early treatment” is the key to saving the lives of patients with breast cancer and improving their quality of life. With the development of molecular biology, the basic and clinical research of breast cancer markers is changing rapidly [1,2,3]. This field play multiple roles in the diagnosis, curative effect observation, follow-up monitoring, and prognosis of breast cancer. Notably, modified nucleosides, which are metabolites of RNA, have great potential in the application of tumor markers [4,5,6,7,8]. Nucleoside differences in healthy adults are very small [9]. Modified nucleosides mainly exist in tRNA and they are obtained from the transcription of RNA and modified by specific methyltransferase and ligase. They are excreted in urine and cannot be reused or degraded by the body. Therefore, the amount of urinary modified nucleosides can reflect the degradation rate of tRNA and can determine the cell proliferation in the body. When the rapid growth of tumor cells in tumor patients accelerates the synthesis of tRNA, the emission of urinary modified nucleosides will increase [10, 11]. In general, urinary modified nucleosides could be used as universal tumor markers for the early diagnosis, treatment, and prognosis of cancer. The urinary levels of 1-methyladenosine (m1A) and 1-methylinosine (m1I) in patients with breast cancer are remarkably higher than those in healthy people [12,13,14,15]. Their emissions are negatively correlated with prognosis. Therefore, m1A and m1I were selected as the research objects in this project to explore their emissions in urine. Urinary modified nucleoside emissions vary greatly at different times of the day and are slightly affected by food. However, the ratio of nucleoside concentration to creatinine (CRE) concentration in random urine and 24-h urine does not change [16]. Therefore, the ratio of modified nucleosides to creatinine (nmol/μmol) was used to reflect the contents of m1A and m1I in urine samples from healthy people and patients with breast cancer. The structures of m1A, m1I, and CRE are shown in Fig. 1.

The structures of 1-methyladenosine, 1-methylinosine and creatinine. a 1-methyladenosine (m1A); b 1-methylinosine (m1I); c Creatinine (CRE)
Various methods to quantify nucleoside using high performance liquid chromatography have been developed and validated. In comparison with other components, the content of nucleoside in the urine with complex chemical composition is very low. Therefore, special pretreatment method should be used for the separation, enrichment, and purification from the complex matrix. The new solid-phase microextraction technology is a good choice, which is easy to operate, fast, and environmentally friendly. Magnetic dispersive solid-phase micro-extraction (MDSPME), a new pretreatment method that uses magnetic or magnetic modified materials as adsorbents [17, 18], has been gradually developed. With large specific surface area, good chemical stability, positive surface charge, and anion exchange function, graphene oxide (GO) is suitable for absorbing nucleotides with negative charge. GO and magnetic materials have the characteristic of easy separation, resulting in a new composite material, which can be used as a high-efficiency magnetic adsorbent [19]. MDSPME does not need the small traditional SPE column, but the adsorbent material is placed directly into the sample solution. As the contact area between the adsorbent and target analyte greatly increase, the extraction efficiency is improved. In addition, the use of external magnetic field for magnetic separation is simple and fast, thus greatly shortens the experimental time. In recent years, the solid-phase microextraction of magnetic graphene oxide (Fe3O4@GO) has been widely used in environmental and food analysis [20,21,22,23,24], but its application in medical field is rarely reported.
In this study, the solid-phase microextraction technology of Fe3O4@GO was established to purify and enrich m1A and m1I in urine samples obtained from patients with breast cancer and healthy patients, and the contents of m1A and m1I were determined by HPLC. In addition, this study aimed to optimize the chromatographic conditions and the adsorption–elution conditions of Fe3O4@GO. This study provides a new idea for the pretreatment and analysis of urinary modified nucleoside as a tumor marker of breast cancer and a new method for the early diagnosis and prognosis tracking of breast cancer for the application of modified nucleoside in clinical practice.
Materials and Methods
Regents and Materials
Creatinine, formic acid, glacial acetic acid, acrylic acid, n-pentanol, and hexanol were obtained from Aladdin Reagent Company (Shanghai, China). 1-methylinosine and 1-methyladenosine were obtained from Research Chemical Reagent Company. (Toronto, Canada). Analytical reagents including ammonium acetate, sodium chloride, sodium hydroxide, ethanol, n-butanol, and hydrochloric acid were obtained from Chinese Medicine Group Chemical Reagent Company. (Shanghai, China). HPLC-grade methanol (MeOH) for liquid chromatography was obtained from Sigma Company. (St., Louis, Mo., USA). All water used in the study were double-distilled water.
Instrumentation
The instruments used include LC-15C high-performance liquid chromatograph (Shimadzu Corporation, Japan), Agilent TC-C18 column (4.6 mm × 250 mm, 5 µm, Agilent Technologies, USA), FTIR spectrometer (Thermo Fisher Scientific, USA), BS124S electronic balance (Beijing Sartorius Instrument System Limited Company, China), SZ-93 Automatic Double Pure Water Distillatory (Shanghai Ya Rong Biochemical Instrument Factory, China), IKA Vortex Genius 3 vortex mixer (IKA- Labortechnik, Germany), X-ray photoelectron spectrometer (Philips, Netherlands), and GEM-2100 transmission electron microscope (JEOL, Netherlands).
Synthesis of Graphene Oxide
The previous synthesis and characterization methods for graphene oxide of the research group were used for reference [25]; the synthesis of GO sheets adopted the Hummer’s method, which is a common preparation method for graphene oxide. Concentrated sulfuric acid (98%) was added into a 1000 mL round bottom flask, and 36 mL phosphoric acid (85%) was added under stirring in ice water bath. After adding 2.7 g graphite powder, 16.2 g potassium permanganate (mass ratio of graphite to potassium permanganate is 1:6) was added for a few times within 10 min, and the solution became dark green. The reaction was stirred in ice water bath (0 ℃) for 4 h, heated to 35 ℃ for 4 h, and finally stirred at 50 ℃ for 21 h. Then, the mixture was continued to be stirred in ice water bath, 360 mL of deionized water was added slowly, and the color of the solution changed from dark green to brownish red with bubbles. Then, approximately 8 mL of hydrogen peroxide was added and the color changed from brown–red to bright yellow. After centrifuging at 6000 rpm for 15 min in a low-speed, large-capacity multi tube centrifuge, 200 mL of 30% HCl was added. Next, the compound was washed neutral with deionized water, washed once with anhydrous ethanol, and dried in vacuum for 24 h.
Synthesis of Fe3O4@GO Nanoparticles
Fe3O4@GO nanoparticles were generated via chemical co-precipitation of Fe2+ and Fe3+ in the presence of GO. In brief, 200 mg GO was added to 200 mL of water, and the dispersed solution was sonicated for 2 h. A total of 11.25 mL of 2 mol/L FeCl3∙6H2O and 3.0 g FeCl2∙4H2O were dissolved in 50 mL of water, and the mixture was added to 1 mg/mL GO solution. Next, NH3∙H2O was added to co-precipitate Fe3O4 at 50 ℃ under argon gas with mechanical agitation until the pH reached 10.0–11.0. The reaction solution was agitated for 3 h. Then, the temperature of reaction solution was elevated to 90 ℃ and maintained for 1.5 h. Next, the reaction solution was left to cool at room temperature. Finally, the black-brown Fe3O4@GO was collected by magnetic separation, washed with water and anhydrous ethanol, and dried under vacuum at 50 ℃ for 24 h.
Characterization of Fe3O4@GO Nanoparticles
The morphology and structure of Fe3O4@GO nanoparticles were characterized by transmission electron microscopy, X-ray diffractometer, and Fourier transform infrared spectroscopy.
Standard Solution Preparation
First, a 10.00 mg/mL creatinine standard reserve solution was prepared and placed in 4 ℃ storage reserve, according to the need to dilute the standard reserve solution water into a certain concentration of standard working solution. Next, m1A and m1I standard reserve solutions of 1.000 and 1.010 mg/mL, respectively, were prepared and placed in frozen storage reserve. The m1A and m1I standard reserve solutions were diluted into a certain concentration of mixed standard working solution.
Collection and Preparation of Urine Sample
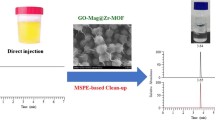
Urine samples were divided into disease and healthy control groups. The urine samples of disease group were obtained from eight untreated patients with breast cancer (A1–A8) from the Quanzhou First Hospital Affiliated to Fujian Medical University. Urine samples of healthy control group were randomly obtained from eight healthy students (B1–B8) from the pharmacy department of Quanzhou First Hospital Affiliated to Fujian Medical University. Following collection, all urine samples were set aside in −20 ℃ sealed preservation immediately. Before analysis, the urine samples were thawed at room temperature. Exactly 10 mL of urine sample was obtained, its pH was adjusted to 10.0 with 0.05 mol/L NaOH solution, the sample was transferred into a 50 mL volumetric flask, and the volume was fixed to the scale for standby. Then, the samples were centrifuged at 5000 rpm for 15 min, and 20 mL of supernatant was collected as the adsorption supply phase reserve. A total of 95.0 mg MGOs were added to 20 mL of the aforesaid solution in 50 mL EP tube. The mixture was dispersed by vortex for 6 min. Then, the MGOs were collected by a magnet, whereas the supernatant liquid was discarded. The collected MGOs were immersed in 2 mL of 0.5% acetic acid aqueous solution saturated by n-butanol and vortexed for 10 s, followed by the separation of the suspension through ultrasound (power 300 W) elution for 5 min at 40 °C. Magnet was used to separate the MGOs again, and the eluent was filtered using a 0.22-µm filter, frozen for 9 h, and then re-dissolved with 80 μL of water. A total of 20 µL of supernatant liquid was injected into the HPLC system for sample determination. Figure 2 illustrates the complete process of MDSPME system combined with HPLC–DAD for the determination of urine samples.

The process of MDSPME system combined with HPLC–DAD for the determination of urine samples
In addition, 50 µL of urine samples were diluted with water for 200–500 times after thawing at room temperature, filtered by 0.22-µm filter, and injected into the HPLC system.
Analysis of Chromatographic Conditions for m1A and m1I
The LC-15C system was used for the chromatographic separation of the aqueous solution. The analytes were separated by Agilent TC-C18 column (4.6 mm × 250 mm, 5 µm) under gradient elution conditions. The mobile phase consisting of (A) 0.03% acetic acid/5 mmol/L-ammonium acetate and (B) MeOH was applied at a flow rate of 1.0 mL/min (gradient elution mode: 0–8.5 min, 4% B; 8.5–14.0 min, 4%–25% B; 14.0–25.0 min, 25% B; 25–30 min, 25%–4% B). The wavelength for detection was 254 nm. The injection volume of the sample solution was 20 µL. The column temperature was controlled at 30 °C.
Analysis of Chromatographic Conditions for Creatinine
Creatinine was separated by Agilent TC-C18 column (4.6 mm × 250 mm, 5 µm) for analysis and detected by LC-15C system directly after diluting. The mobile phase including 95% of (A) 0.05% acetic acid/30 mmol/L ammonium acetate and 5% of (B) MeOH was applied at a flow rate of 0.9 mL/min. The wavelength for detection was 254 nm. The injection volume of the sample solution was 20 µL. The column temperature was controlled at 30 ℃.
Results and Discussion
Characterization of Fe3O4@GO
Figure 3a shows the FTIR spectra of Fe3O @GO, GO, and Fe3O4 nanoparticles. The comparison of several characteristic absorption peaks shows that Fe3O4 nanoparticles have been successfully prepared. Figure 3b indicates the XRD patterns of Fe3O4@GO. Fe3O4 displayed six notable diffraction peaks (2θ = 30.2°, 35.5°, 43.6°, 53.4°, 57.3°, and 62.9°), which were assigned to the crystal faces of (220), (311), (400), (422), (511), and (440), respectively. Figure 3c shows the TEM image of GO loaded with Fe3O4 nanoparticles. The Fe3O4 nanoparticles are assembled on the GO sheet to form composite materials. Figure 3d indicates that the TGA curves of Fe3O4@GO, which were obtained by heating up to 800 ℃ under N2, exhibiting the amount of GO bound onto the surface of Fe3O4 and thermal stability of Fe3O4@GO. The characterization results showed that Fe3O4@GO was successfully synthesized.

Characterization of Fe3O4@GO nanoparticles: a FTIR, b XRD, c TEM at 50 nm and at 20 nm, d TGA
Optimization of Extraction Conditions
The efficiency of extraction procedure was affected by several factors, including adsorption time, pH, ionic strength, mass of adsorbent, elution solvent, ultrasonic time, and ultrasonic temperature.
Optimization of Adsorption Time
A vortex oscillator, which could shorten the diffusion time of adsorbent through a unique vortex system was used to make Fe3O4@GO fully mix with the target and absorb it. The experiments were actualized by mixing 30 mL of nucleotide solution (0.5 µg/mL, pH = 10) with 95.0 mg MGO in a series of vortex time ranging from 2 to 7 min. The experimental results showed that with the increase of vortex time, the adsorption effect of the target could be improved, and the extraction recovery of the remaining target in the adsorption solution was almost unchanged after 5 min (Supporting Information, Fig. S1A). Consequently, to ensure the adsorption effect, the practical choice for vortex time is 6 min.
Optimization of pH
Adsorption pH can change the surface charge density of Fe3O4@GO, which is an important factor affecting the adsorption effect. Therefore, this study investigated the adsorption effect of the adsorption solution pH in the range of 3.0–12.0 under 6 min of vortex, whereas 95 mg MGO was mixed with nucleotide mixture (0.5 µg/mL). The experimental results showed that the adsorption effect of m1I was almost unaffected by the pH, while the adsorption effect of m1A was the best at 10.0. (Supporting Information, Fig. S1B). Therefore, a mixed solution of nucleotides with a pH of 10 was selected for the next study.
Optimization of Ionic Strength
The experiment about the effect of ionic strength by adding different concentrations of NaCl (0.1, 0.2, 0.3, 0.4, and 0.5 M) to the mixed solution of nucleotides were investigated further. The results showed that the adsorption efficiency of m1I did not change obviously with the increase of ionic strength, but the adsorption efficiency of m1A decreased (Supporting Information, Fig. S1C). This result was obtained because of the existence of electrostatic adsorption and the competition between NaCl and the target substance on Fe3O4@GO that inhibited the adsorption of the target substance. Therefore, the following experiments were performed without adding NaCl.
Optimization of MGO Mass
After determining the adsorption time, adsorption liquid pH, and NaCl concentration, the amount of adsorbent used was increased to increase the adsorption rate of the target. Thus, the experiment determined the effects of MGO mass by working through different nano-sorbent doses (20–100 mg) mixed with 30 mL of nucleotide mixture (0.5 µg/mL, pH = 10.0) under 6 min vortex. The results showed that with the increase of MGO mass, the adsorption effect increased, and the target was almost completely adsorbed as the amount of absorbent exceeded 80 mg (Fig. 4). Moreover, MGO absorbed a small amount of impurities because of the similar structure of the components in the urine samples. To ensure that the target was completely adsorbed, we selected 95 mg MGO for the following experiments.

The effect of MGO mass on adsorption efficiency
Optimization of Elution Solvent
First, we determined that the adsorption effect on m1A was better when the pH of the adsorption solution is alkaline. Thus, different kinds of acid solvents, such as hydrochloric acid, acetic acid, formic acid, and benzoborate acid, at different concentrations were used to desorb the target in this part. The experimental results showed that the elution effect of 0.5% acetic acid aqueous solution on m1A was the best, whereas the elution effect on m1I was poor. To improve the solubility of m1I, we used different proportions of methanol water solution, ethanol water solution, saturated water solution of n-butanol, saturated water solution of n-pentanol, and saturated water solution of n-hexanol. The experimental results indicated that the saturated water solution of n-butanol had the best elution effect on m1I but not on m1A, and 0.5% acetic acid solution saturated with n-butanol as the eluent was selected to elute the two targets to achieve the best elution effect of the two targets at the same time.
Then, the volume of 0.5% acetic acid solution saturated with n-butanol was optimized. When 2 mL of 0.5% acetic acid solution saturated with n-butanol was added (added in twice), m1I and m1A were eluted out.
Optimization of Ultrasonic Time and Ultrasonic Temperature
The cavitation effect of ultrasound makes the target adsorbed on MGO surface in the cavitation field, obtaining the energy of shock wave, thus reducing the adsorption rate. Moreover, the target can convert the absorbed energy into heat energy, thus increasing the solvent temperature and enhancing the dissolution rate of the target. However, if the temperature is very high, the stability of the target is affected. In this experiment, the influence of ultrasonic elution time within 0–15 min on the target determination was investigated. When the ultrasonic time exceeded 5 min, the elution efficiency achieved equilibrium (Supporting Information, Fig. S2A). Therefore, the ultrasonic time was set to 5 min for the following experiments.
In addition, a proper temperature will influence the elution efficiency. The temperature was set to 25, 30, 40, 50, and 60 °C to reveal the effect of ultrasonic extraction temperature on the target determination. The results showed that with the increase of temperature, the elution efficiency increased obviously, but when the temperature reached 40 ℃, the elution efficiency decreased obviously (Supporting Information, Fig. S2B). This may be attributed to the disruption of the hydrogen bonding effect between the analyte and the eluting solvent as the temperature increases, thus causing a decrease in elution efficiency. Therefore, 40 °C was selected for the following experiments.
In conclusion, the optimum extraction conditions for the target in urine samples were addition of 95 mg MGO to the adsorbed solution (with no salt, pH = 10.0) and vortex time of 6 min. Then, 2 mL of 0.5% acetic acid aqueous solution saturated by n-butanol was added to MGO and ultrasonicated for 5 min at 40 ℃.
Reusability of the Nanoparticles
The reusability of the Fe3O4@GO nanoparticles was evaluated for 12 sequential cycles. The Fe3O4@GO nanoparticles could be used for 10 cycles, and the extraction recovery of m1A and m1I were both greater than 90% (Supporting Information, Fig. S3). These indicated a good reusability of the nanoparticles.
Method Validation
This validation was performed following the International Coordinating Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline [26].
After establishing the determination method, the method was validated on urine specimens in terms of relevant parameters of linearity range, limit of detection (LOD), limit of quantification (LOQ), repeatability, stability, intra-day and inter-day precision, and the recovery of extraction.
After preparing a series of mixed standard working solution of m1A and m1I in the range of 1–1000 ng/mL, the sample was injected for analysis after extraction under the optimal conditions. Based on the data obtained, the calibration curves showed good linearity within a specific linear range. Meanwhile, a series of creatinine standard working solution with concentrations of 0–10 μg/mL were prepared, and relevant data were obtained by direct injection analysis. The results indicated that the linear relationship of creatinine was good in the linear range. In addition, the sensitivity was evaluated based on the LODs and LOQs, which were estimated at a signal-to-noise ratio of 3:1 and 10:1, and detailed information is listed in Table 1. The lower limit of quantification samples was tested for 12 consecutive times. The deviations between the tested values and theoretical values, as well as the relative standard deviations (RSDs) of the test results, were calculated. It was found that the deviations between the tested values and theoretical values of m1A and m1I ranged from 4.75 to 7.05%, and the RSDs of the tested parallel samples ranged from 6.75 to 7.16%.
The repeatability of sample preparation was determined by six-replicate experiments, and the RSD % (n = 6) of the sample solutions was <2.4%. The stability of sample solutions was achieved via continuous analysis of the same sample at standing times (0, 2, 4, 6, 12, and 24 h), and the RSD % (n = 6) was <1.6%.
The intra-day and inter-day repeatability was analyzed to investigate the precision of the developed method. The intra-day repeatability was investigated at different concentrations (2, 400, and 750 ng/mL) with 6 replicates daily, and the obtained RSD values ranged from 2.78 to 3.92%. The inter-day repeatability was evaluated for 3 consecutive days, and the RSD values ranged from 3.72 to 4.08%.
The recovery experiment was used to evaluate the reliability of the experimental method. At high, medium, and low levels, a series of appropriate amount of mixed standard solutions (m1A, m1I, and CRE) were added into nine urine samples of the healthy control group. The mixture was extracted under the optimum extraction conditions and determined thrice in parallel by HPLC–DAD. In addition, creatinine recovery rate was directly determined by the mixture solution according to the previous detection conditions. Table 2 lists the recoveries of two modified nucleosides and creatinine in human urine and their corresponding RSD% (n = 3).
Enrichment Factor
The extraction was carried out under the above optimal conditions. EF is defined as the ratio of the analyte concentration elution solution (CR) to its initial concentration (C0) without extraction step. EF was calculated according to Eq. (1). Table 1 shows the concrete values of EF. The EF values of m1A and m1I were 108 and 134, respectively.
Application to Actual Urine Samples
In this assay, the aforesaid optimized MDSPME–HPLC combined method was developed for the quantitative analysis of m1A and m1I in selected human urine samples. Figure 5a shows the HPLC chromatograms obtained for the mixed standard working solution of m1A and m1I with a concentration of 1 μg/mL before and after enrichment. The figure shows that the retention times of m1A and m1I were 7.494 and 18.286 min. Figure 5b shows the HPLC chromatograms, including A1 samples before and after extraction. By comparing the chromatograms of the sample with the standard chromatograms, the components of m1A and m1I can be identified according to the retention time.

The HPLC chromatograms obtained for the nucleotides of mixed standard working and actual samples before and after extraction. a-A contained 1 µg/mL of the modified nucleotides after extraction; a-B contained 1 µg/mL of the modified nucleotides before extraction; b-A the sample of A1 after extraction; b-B the sample of A1 before extraction
Considering the complex matrix in urine, the elution procedure used to detect the two modified nucleosides cannot separate creatinine from other impurities, and another elution procedure was used to detect creatinine. Figure 6 shows the HPLC chromatograms obtained for the standard working solution of creatinine of 5 μg/mL before and after extraction. As shown in Fig. 6, no change was observed in the chromatographic peak area of creatinine before and after extraction. Fe3O4@GO had no adsorption effect on creatinine. Hence, the concentration of creatinine could be determined directly with the urine sample before extraction (Fig. 7).

The HPLC chromatograms obtained for the standard working solution of creatinine before and after extraction. a contained 5 µg/mL of the creatinine after extraction; b contained 5 µg/mL of the creatinine before extraction

The HPLC chromatogram obtained for actual samples before extraction
In this experiment, 15 mL urine were obtained from volunteers in the disease (A) and healthy control groups (B), and the samples were extracted and analyzed according to the previous steps. Each urine sample was measured in parallel thrice, and the average value was obtained as the measurement result. Similarly, the average content of creatinine was considered as the measurement result after three measurements in parallel with each urine sample without extraction. The contents of m1A, m1I, and CRE in the urine of 8 patients with breast cancer were 4.17–14.20, 3.26–13.41, and 5.42–16.82 μmol/mL, respectively, while those in eight healthy women were 1.33–4.95, 1.84–4.11, and 4.51–10.18 μmol/mL, respectively. Urinary modified nucleoside emissions vary greatly at different times of the day and are affected little by food. However, the ratio of urinary modified nucleoside to creatinine (CRE) was constant. Therefore, the ratio of modified nucleosides to creatinine (nmol/μmol) was used to reflect the contents of m1A and m1I in the urine samples of normal people and patients with breast cancer. The ratio of urinary modified nucleoside to creatinine was obtained. The results are summarized in Table 3. The SPSS statistical software package (version 18.0) was used to compare the two results using t test (P < 0.01, two-sided Student’s t test) and analyze their significant difference. By contrast, the contents of m1A/CRE (P = 0.00004) and m1I/CRE (P = 0.00006) in the urine samples of patients with breast cancer patients were significantly higher than those of healthy female volunteers.
Comparison with Literature Methods
The presented method was compared with previous methods for the determination of m1A, and the results are summarized in Table 4. The LOD obtained by the present method was lower than other reported methods (except for the UHPLC-UniSpay-MS/MS method that requires expensive instruments and professional personnel). With this method, two modified nucleosides can be enriched 108–134 times in a relatively short time (11 min), and the results are accurate and reliable. Additionally, the adsorbent nanoparticles can be reused at least 10 times, which can save time and cost for analysis. Taken together, the developed MDSPME-HPLC method is a fast, easy-to-operate, and cost-efficient method for detecting m1A and m1I.
Conclusion
This study developed a novel analytical method of MDSPME combined with HPLC for the determination of m1A and m1I in human urine. The Fe3O4@GO nanoparticles is a MDSPME adsorbent characterized by large specific surface area, strong magnetism, high adsorption capacity, and good reusability. This magnetic extraction avoided the time-consuming column passing or filtration step and simplified sample preparation procedures, which can greatly improve the extraction rate. The complete extraction could be performed in 11 min, with high extraction recovery. The developed MDSPME-HPLC method can be successfully applied in m1A and m1I analysis in human urine. This new approach not only had high detection sensitivity and precision but also produced great recoveries in intricate sample matrices. Compared with other methods for determining m1A and m1I, the proposed method showed the advantages of simplicity, efficiency, cost-efficient, and environmental protection. This method can provide a new method for the pretreatment and analysis of urinary modified nucleoside as a tumor marker of breast cancer and provide a new idea for the early non-invasive diagnosis and prognosis tracking of breast cancer. In the future, functionalization modification can be carried out on the magnetic adsorbed nanoparticles, which can further enhance its adsorption capacity for analyzing more complex biological samples.
Data Availability
No datasets were generated or analyzed during the current study.
References
Li J, Liu L, Feng Z, Wang X, Huang Y, Dai H, Zhang L, Song F, Wang D, Zhang P, Ma B, Li H, Zheng H, Song F, Chen K (2020) Tumor markers CA15-3, CA125, CEA and breast cancer survival by molecular subtype: a cohort study. Breast Cancer 27:621–630
Masuda T, Ueo H, Kai Y, Noda M, Hu Q, Sato K, Fujii A, Hayashi N, Tsuruda Y, Otsu H, Kuroda Y, Eguchi H, Ohno S, Mimori K, Ueo H (2020) N-cadherin mRNA levels in peripheral blood could be a potential indicator of new metastases in breast cancer: a pilot study. Int J Mol Sci 21:511
Tarighati E, Keivan H, Mahani HA-O (2023) A review of prognostic and predictive biomarkers in breast cancer. Clin Exp Med 23:1–16
Iwanowska A, Yusa S, Nowakowska M, Szczubiałka K (2016) Selective adsorption of modified nucleoside cancer biomarkers by hybrid molecularly imprinted adsorbents. J Sep Sci 39:3072–3080
Lin X, Wang L, Lin H, Lin D, Lin J, Liu X, Qiu S, Xu Y, Chen G, Feng S (2019) A novel urine analysis technique combining affinity chromatography with Au nanoparticle based surface enhanced Raman spectroscopy for potential applications in non-invasive cancer screening. J Biophotonics 12:e201800327
Godoy AT, Eberlin MN, Simionato AVC (2020) Targeted metabolomics: liquid chromatography coupled to mass spectrometry method development and validation for the identification and quantitation of modified nucleosides as putative cancer biomarkers. Talanta 210:120640
Huang H, Weng H, Chen J (2020) m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37:270–288
Mohl DA, Lagies S, Zodel K, Zumkeller M, Peighambari A, Ganner A, Plattner DA, Neumann-Haefelin E, Adlesic M, Frew IJ, Kammerer B (2023) Integrated metabolomic and transcriptomic analysis of modified nucleosides for biomarker discovery in clear cell renal cell carcinoma. Cells 12:1102
Lu Z, Wang Q, Wang M, Fu S, Zhang Q, Zhang Z, Zhao H, Liu Y, Huang Z, Xie Z, Yu H, Gao X (2017) Using UHPLC Q-Trap/MS as a complementary technique to in-depth mine UPLC Q-TOF/MS data for identifying modified nucleosides in urine. J Chromatogr B Analyt Technol Biomed Life Sci 1051:108–117
Jiang Y, Ma Y (2009) A fast capillary electrophoresis method for separation and quantification of modified nucleosides in urinary samples. Anal Chem 81:6474–6480
Lagies S, Schlimpert M, Braun LM, Kather M, Plagge J, Erbes T, Wittel UA, Kammerer B (2019) Unraveling altered RNA metabolism in pancreatic cancer cells by liquid-chromatography coupling to ion mobility mass spectrometry. Anal Bioanal Chem 411:6319–6328
Struck W, Waszczuk-Jankowska M, Kaliszan R, Markuszewski MJ (2011) The state-of-the-art determination of urinary nucleosides using chromatographic techniques “hyphenated” with advanced bioinformatic methods. Anal Bioanal Chem 401:2039–2050
Feng S, Zheng Z, Xu Y, Lin J, Chen G, Weng C, Lin D, Qiu S, Cheng M, Huang Z, Wang L, Chen R, Xie S, Zeng H (2017) A noninvasive cancer detection strategy based on gold nanoparticle surface-enhanced raman spectroscopy of urinary modified nucleosides isolated by affinity chromatography. Biosens Bioelectron 91:616–622
Seidel A, Brunner S, Seidel P, Fritz GI, Herbarth O (2006) Modified nucleosides: an accurate tumour marker for clinical diagnosis of cancer, early detection and therapy control. Br J Cancer 94:1726–1733
Willmann L, Erbes T, Halbach S, Brummer T, Jäger M, Hirschfeld M, Fehm T, Neubauer H, Stickeler E, Kammerer B (2015) Exometabolom analysis of breast cancer cell lines: Metabolic signature. Sci Rep 5:13374
Zamora-Ros R, Rabassa M, Cherubini A, Urpi-Sarda M, Llorach R, Bandinelli S, Ferrucci L, Andres-Lacueva C (2011) Comparison of 24-h volume and creatinine-corrected total urinary polyphenol as a biomarker of total dietary polyphenols in the Invecchiare InCHIANTI study. Anal Chim Acta 704:110–115
Ghorbani M, Chamsaz M, Aghamohammadhasan M, Shams A (2018) Ultrasonic assisted magnetic dispersive solid phase microextraction for pre concentration of serotonin-norepinephrine reuptake inhibitor drugs. Anal Biochem 551:7–18
Nazario CE, Fumes BH, da Silva MR, Lanças FM (2017) New materials for sample preparation techniques in bioanalysis. J Chromatogr B Analyt Technol Biomed Life Sci 1043:81–95
Tang HZ, Wang YH, Li S, Wu J, Li JW, Zhou HY, Gao ZX (2019) Graphene oxide composites for magnetic solid-phase extraction of twelve quinolones in water samples followed by MALDI-TOF MS. Anal Bioanal Chem 411:7039–7049
Zhang HF, Shi YP (2012) Magnetic retrieval of chitosan: extraction of bioactive constituents from green tea beverage samples. Analyst 137:910–916
Moradi Z, Alipanahpour Dil E, Asfaram A (2019) Dispersive micro-solid phase extraction based on Fe(3)O(4)@SiO(2)@Ti-MOF as a magnetic nanocomposite sorbent for the trace analysis of caffeic acid in the medical extracts of plants and water samples prior to HPLC-UV analysis. Analyst 144:4351–4361
Zhao B, Wu H, Liu Y, Tian X, Huo Y, Guan SJAM (2019) Magnetic solid-phase extraction based on g-C3N4/Fe3O4/MoS2 as a magnetic adsorbent for HPLC-UV determination of fluoroquinolones in chicken and eggs. Anal Methods 11:1491–1499
Li N, Chen J, Shi YP (2019) Magnetic nitrogen-doped reduced graphene oxide as a novel magnetic solid-phase extraction adsorbent for the separation of bisphenol endocrine disruptors in carbonated beverages. Talanta 201:194–203
Dil EA, Doustimotlagh AH, Javadian H, Asfaram A, Ghaedi M (2021) Nano-sized Fe(3)O(4)@SiO(2)-molecular imprinted polymer as a sorbent for dispersive solid-phase microextraction of melatonin in the methanolic extract of Portulaca oleracea, biological, and water samples. Talanta 221:121620
Chen X, Wu Y, Huang L, Yang L, Hong R, Yao H, Li S (2019) Magnetic dispersive solid-phase micro-extraction combined with high-performance liquid chromatography for determining nucleotides in Anoectochilus roxburghii (Wall.) Lindl. J Pharm Biomed Anal 174:432–440
ICH (2022) M10 Bioanalytical method validation and study sample analysis. Guidance for Industry Final version
Kogaki T, Ohshio I, Ura H et al (2021) Development of a highly sensitive method for the quantitative analysis of modified nucleosides using UHPLC-UniSpray-MS/MS. J Pharm Biomed Anal 197:113943
Omran MM, Rashed RE, Darwish H, Belal AA, Mohamed FZ (2020) Development of a gas chromatography-mass spectrometry method for breast cancer diagnosis based on nucleoside metabolomes 1-methyl adenosine, 1-methylguanosine and 8-hydroxy-2’-deoxyguanosine. Biomed Chromatogr 34(1):e4713
Raćkowska E, Bobrowska-Korczak B, Giebułtowicz J (2019) Development and validation of a rapid LC-MS/MS method for determination of methylated nucleosides and nucleobases in urine. J Chromatogr B Analyt Technol Biomed Life Sci 1128:121775
Cho SH, Choi MH, Lee WY, Chung BC (2009) Evaluation of urinary nucleosides in breast cancer patients before and after tumor removal. Clin Biochem 42(6):540–543
Hsu WY, Chen WT, Lin WD et al (2009) Analysis of urinary nucleosides as potential tumor markers in human colorectal cancer by high performance liquid chromatography/electrospray ionization tandem mass spectrometry. Clin Chim Acta 402(1–2):31–37
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
HL performed the experiment part; HL and MH wrote the main manuscript; HX analyzed the experimental data; FX completed Fig. 2; LY reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no financial and personal relationships with other individuals or organizations that would inappropriately influence this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hong, L., Mao, H., Huang, X. et al. Integration of Magnetic Dispersive Solid-Phase Micro-extraction with HPLC for Precise Detection of Tumor Markers 1-Methyladenosine and 1-Methylinosine in Urine Samples from Breast Cancer Patients. Chromatographia 87, 407–418 (2024). https://doi.org/10.1007/s10337-024-04334-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-024-04334-y