
Abstract
For the extraction and measurement of plasma amino acids using LC/ESI–MS/MS technology, a straightforward and dependable approach has been established. The approach employs a small amount of sample and fully quantifies it using a labeled internal standard of amino acids. Water, formic acid, and methanol were used as the mobile phase in a gradient method to obtain clear separation. On a Shimatzu mass spectrometer 8030, optimized multiple reaction monitoring (MRM) was utilized to find amino acids. All amino acids have coefficients of correlation that range from 0.91 to 0.99, and they are all linear over their respective reference ranges. Precision’s intra-day and inter-day coefficients of variation (CV %) ranged from 3.29 to 11.73% and 5.04 to 12.48%, respectively. Amino acid recovery ranged from 92.1 to 108.2%.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The analysis of a person’s plasma amino acid profile has become more important for determining their nutritional condition. Estimating amino acids is helpful in clinical research of hereditary metabolic abnormalities in both newborns and adults [1]. To check on a person’s health status, it is now a common test in diagnostic laboratory [2]. The patient’s entire metabolic profile is examined based on the clinical findings, and a treatment plan is created to meet his health needs. Either amino acid supplements or dietary adjustments, such as increasing protein intake, are used as treatments. Additionally, periodic clinical tests are used to monitor amino acids profiling and assess health condition. One such ailment is maple syrup urine disorder (MSUD), where the effectiveness of treatment for the amino acid profile is periodically assessed.
The classic methods for estimating amino acids include HPLC, GCMS, etc. Ion exchange chromatography is a technology that is frequently used to separate plasma amino acids, although it has longer runtimes and is, therefore, difficult to use in commercial application [3]. Other HPLC techniques offer the benefit of shorter run times, although it might be challenging to separate some clinically important amino acids, such as isomers of leucine [4,5,6]. Ninhydrin post-column derivatization is also used. Thus, the test's time and financial costs mount up. The test method becomes cumbersome as a result. Pre-column derivatization was another extensively used HPLC technique. Some of the chemicals that have been employed as derivatizing agents include phenylisothiocyanate and pthalaldehyde [7,8,9].
The goal of this experiment was to create an easy-to-replicate technique for extracting plasma amino acids. Less time should be spent running the method, and it should be reliable for detecting and quantifying analytes with less interference.
The current approach takes 6 min to examine 29 amino acids. The C18 column is used for separation based on the liquid chromatography principle, and mass spectrometry is used for quantification with a deuterium-labeled internal standard. The approach is straightforward because analytes do not need to be derivatized before or after the column. The material, which was extracted with methanol, is stable for 24 h at 4 °C and is used for testing.
Materials and Method
Chemicals and Reagents
We purchased, hydrazine monohydrate (98%), glycine, alanine, valine, leucine, methionine, phenylalanine, tyrosine, aspartic acid, glutamic acid, ornithine, citrulline, arginine, and L-carnitine from Sigma-Aldrich; isotopically labelled amino acid and internal standards from Cambridge Isotope Laboratories. All other chemical and solvents were of the highest purity available from commercial sources and used without further purification.
LC–MS/MS Analysis of Amino Acids
The chromatographic separation for plasma was carried out by Shimadzu LCMS 8030 (Shimadzu, Kyoto, Japan) instrument. It is equipped with binary pump, auto sampler, column oven, ESI probe, and UF-sweeper enabled triple quadrupole mass spectrometer. The system was managed by Lab Solutions software version 5.60 SP2. Argon gas was purchased with 99.9% pure. Nitrogen generator was purchased from Peak Scientific. Nitrogen gas was used as drying gas and nebulizing gas. HPLC system used two mobile phases for amino acids separation. Mobile phase A was 0.1% formic acid made in water. Mobile phase B was 100% methanol. Rinsing solution used consisted of 60:40 methanol–water mixture. All solvents were sonicated and filtered through 0.2-µm filter. C18 column of Phenomenex make was used for amino acids separation. Column had 2.6µ particle size and 100 mm × 3 mm id. Total flow rate was 0.2 mL·min-1 throughout the run. From 0 to 4.5 min, concentration of phase B was 3%. From 4.5 to 5.1 min, it was 90% and from 5.1 to 6th minute, it was kept 3%. System was equilibrated to 0.2 mL·min-1 of flow rate keeping mobile phase B concentration 3% for 30 min before starting the analysis.
Standard Preparation
All amino acids standard were prepared in de-ionized water except tryptophan, glutamic acid, and tyrosine. They were prepared in 100 mmol·L-1 of HCl and stored at 4˚C. EDTA plasma samples were collected from healthy individuals. Their age, gender, and other clinical information were recorded. Written consent was obtained from all subjects. Samples were stored at 4°C.
Sample Preparation
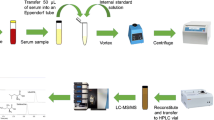
10 µL of freshly drawn plasma sample (collected in EDTA tube) was dispensed in 2 mL micro-centrifuge tube. 200 µL working internal standard mixture of methanol:hydrate hydrazine:oxalic acid:acetonitrile:water containing stable isotopically labeled internal standards and 10 µL of dithiothreitol (500 mM) were added to it [10, 11]. Tube was vortexed and incubated at room temperature for 20 min. Sample was further centrifuged at 10,000 rpm for 10 min and 20 µL supernatant was used for further analysis.
Validation Study
Stock mixture of 29 amino acids was made and serially diluted further to obtain 6 standards covering biological reference range of individual amino acid. Linearity study was performed for every amino acid by plotting standard curve for amino acid concentration (µmol·L-1) vs. response in area. The correlation coefficient value for every amino acid was > 0.90. For precision study, two concentrations were selected for each amino acid based on their biological reference range—one covering normal reference range and other above the upper control limit of reference range. These two levels were run five times within a day and over 5 days. The intra-assay coefficient of variation (CV) was between 1 and 7% as determined by replicate analysis. The inter-assay CV was between 2 and 12% as determined by replicate analysis of the same quality control plasma sample stored at –70 °C and analyzed for 5 days. The recovery of the plasma amino acids assay was assessed following NCCLS approved guidelines EP6-A, evaluation of quantitative measurement procedures: a statistical approach 2003. The recovery of the plasma amino acids was between 91 and 104% as determined by mixing each amino acid standard separately in a pooled plasma sample, and recovery was calculated as the difference between spiked and unspiked plasma samples (Fig. 1).

LC and MS spectra of separation of plasma amino acids over 6 min of run-time by LC/ESI–MS/MS technology. Response is shown in terms of ion currents corresponding to the product ions of respective analytes obtained in MRM mode
Results and Discussion
Validation
Plasma amino acids MRM were optimized on LCMS 8030 for fine tuning of mass transitions and MS voltages. Optimization is done to achieve best possible response of analyte (Table 1). It involves fine tuning of precursor and product ion required in MRM transitions (multiple reaction mode).
In the plasma, the homocysteine-related metabolites were initially not detected, since homocysteine was lost during sample clean up. To overcome this problem, DTT was added before the precipitation step in internal standard mixture and the samples were reanalyzed. In aqueous solution, DTT exists as equilibrium mixture of the oxidized to reduced form and the quantitation tools.
Linearity studies were done to evaluate the analytical measurement range of each amino acid. As mentioned in Table 2, the linearity of the calibration curve of each individual AA ranged from 0.78 μmol·L−1 to 500 μmol·L−1. The method was linear within allowable systematic error of 10% up to 500 nmol·L−1 for all the amino acids studied. The r2 of the calibration curve (Table 2) was the mean r2 of the calibration curve measured at 3 independent days performed on amino acids. Linearity study was performed for every amino acid by plotting standard curve for amino acid concentration (µmol·L−1) vs. response in area. The correlation coefficient value for every amino acid was ranging from 0.9 to 1.0
This method’s performance characteristics are demonstrated in Table 2. Analyzing QC samples allowed researchers to identify the repeatability and reproducibility variances for the quantitative amino acids. All amino acids that were studied had within-run (intra-day) CVs that ranged from 3.29 to 11.73. For each of the examined amino acids, the CV ranged from 5.04 to 12.48% between runs (inter-day).
Following EP-24-A recommendations approved by the NCCLS and CFI guidelines for bioanalytical procedures, the accuracy of the plasma amino acids assay was evaluated. Table 2 displays the preparation of 29 plasma samples spiked with known amino acids. An in-lab TMS assay was used to evaluate the samples. For aspartic acid and phenyl alanine, respectively, the accuracy varied from 95.2 to 108.1%.
The computed LOD (0.01–0.2 mol·L−1) and LOQ (0.001–0.1 mol·L−1) were low enough to diagnose individuals with an IEM of AA metabolism and analyze a person’s nutritional status, meeting the requirements of analytical sensitivity.
The current approach for measuring amino acids in plasma is straightforward in terms of sample processing. For compressive profiles, previously reported methods either had pre-column or analytical derivatization such alkylation [12, 13]. However, the current technique does not call for derivatization at the pre-column or post-column stages, which reduces both processing time and cost. The basis for amino acid detection is the mass spectrometry principle. Therefore, a little change in the retention time (RT) due to a change in the mobile phase lot or any other external cause will have no effect on the analysis. Homocysteine was the hardest to detect. However, because it is a cardiac marker, its diagnosis is crucial for amino acid profiling. DTT was used extensively in the extraction and measurement of plasma homocysteine [13]. Mobile phases are easy to prepare and not pH dependent. Overall, reagent preparation is not time consuming or complex. The method evaluates the most amino acids in a shorter amount of time than the 18-min method that was previously published [12].
The amino acids that are assessed using this technology have a significant clinical impact on nutritional research on inborn errors of metabolism as well as prediabetes markers. Plasma amino acids are specifically connected with insulin resistance [14,15,16], the prediction of the onset of diabetes [17, 18], and the results of intervention [19,20,21] in a number of recent investigations. A suitable linear response and good precision are shown by the validation investigation over the analytes anticipated reference range.
As a result, the technique may be useful for commercial diagnostic application. In contrast to previous tandem mass spectrometric approaches, no shift in retention times has been seen in more than 10,000 plasma amino acid runs that have been carried out in our lab [12, 22, 23].
Conclusion
In conclusion, this technique has been designed and tested to be reliable and robust for absolute quantification of 29 amino acids in plasma. The approach only has a 6-min analysis time. It was discovered that this approach is reliable for the physiologically significant range of 0–500 µM·L-1. There was no RT drift for any amino acid, and the chromatography was reliable and reproducible. Present data demonstrate best alternative to traditional GC–MS-based or derivatisation-based HPLC method.
Data availability
The data that support the findings of this study are available on request from the corresponding author (shrimant.p@gd-lab.com).
Abbreviations
- HPLC:
-
High-performance liquid chromatography
- GCMS:
-
Gas chromatography–mass spectrometry
- IEM:
-
Inborn error of metabolism
- TMS:
-
Tandem mass spectrometry
- ESI:
-
Electrospray ionization
- µL:
-
Micro liter
- µM·L-1 :
-
Micromole per liter
- MRM:
-
Multiple reaction monitoring
- RT:
-
Retention time
References
Michael A, Karen J, Nichole AR (2007) Analysis of 25 underivatized amino acids in human plasma using ion-pairing reversed-phase liquid chromatography/time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 21:2717–2726
Suresh B, Shareef M, PavanKumar A, Taranath K (2002) HPLC method for amino acids profile in biological fluids and inborn metabolic disorders of aminoacidopathies. Indian J Clin Biochem 17(2):7–26
Elisabeth L, William L, Marzia P (2005) Analysis of plasma amino acids by HPLC with photodiode array and fluorescence detection. Clin Chim Acta 354:83–90
Robitaille L, Hoffer LJ (1988) Measurement of branched chain amino acids in blood plasma by high performance liquid chromatography. Can J Physiol Pharmacol 66:613–617
Schadewaldt P, Bodner-Leidecker A, Hammen HW, Wendel U (1999) Significance of l-alloisoleucine in plasma for diagnosis of maple syrup urine disease. Clin Chem 45:1734–1740
Schadewaldt P, Bodner-Leidecker A, Hammen HW, Wendel U (2000) Formation of l-alloisoleucine in vivo: an l-[13C] isoleucine study in man. Pediatr Res 47:271–277
Tom T, Paul L, Alexander H (1994) Plasma amino acids determined by liquid chromatography within 17 minutes. Clin Chem 40(2):245–249
Monique P, Christine V, Konstantinos P, Claire E, Jean-Paul S, Denis B (2005) Ion-pairing reversed-phase liquid chromatography/electrospray ionization mass spectrometric analysis of 76 underivatized amino acids of biological interest: a new tool for the diagnosis of inherited disorders of amino acid metabolism. Rapid Commun Mass Spectrom 19:1587–1602
Michael P, Robert W (2007) Simple and rapid quantitative high-performance liquid chromatographic analysis of plasma amino acids. J Chromatogr B Analyt Technol Biomed Life Sci 852(1–2):646–649
Shrimant P, Mahesh H, Susheel S, Akshata S, Pallavi K (2019) Fast & reliable liquid chromatography mass spectrometry method for determination of amino acids & acylcarnitines including succinyl acetone in dried blood spots for newborn screening. Int J Med Sci 6(7):1–11
Mahesh H, Pramod I, Shrimant P, Ashwini Y (2015) Two tier analysis of organic acid disorders: a comprehensive approach for newborn screening. Int J of Biomed Adv Res 6(02):84–90
Prinsen HCMT, Schiebergen-Bronkhorst BGM, Roeleveld MW, Jans JJM, Sain-van M, Visser G, van Hasselt PM, Verhoeven-Duif NM (2016) Rapid quantification of underivatised amino acids in plasma by hydrophilic interaction liquid chromatography (HILIC) coupled with tandem mass-spectrometry. J Inherit Metab Dis 39:651–660
Hariharan M, Sundar N, Ted V (1993) Systematic approach to the development of plasma amino acid analysis by high-performance liquid chromatography with ultraviolet detection with precolumn derivatization using phenyl isothiocyanate. J Chromatogr 621:15–22
Huffman K, Shah S, Stevens R, Bain JR, Muehlbauer M (2009) Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care 32:1678–1683
Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD et al (2009) A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9:311–326
Tai ES, Tan ML, Stevens RD, Low YL, Muehlbauer MJ et al (2010) Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 53:757–767
Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP (2011) Metabolite profiles and the risk of developing diabetes. Nat Med 17:448–453
Wurtz P, Soininen P, Kangas AJ, Ronnemaa T, Lehtimaki T (2013) Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 36:648–655
Laferrere B, Reilly D, Arias S, Swerdlow N, Gorroochurn P (2011) Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci Transl Med 3:80–82
Hsiao G, Chapman J, Ofrecio JM, Wilkes J, Resnik JL (2011) Multi-tissue, selective PPARgamma modulation of insulin sensitivity and metabolic pathways in obese rats. Am J Physiol Endocrinol Metab 300:E164-174
Shah SH, Crosslin DR, Haynes CS, Nelson S, Turer CB (2012) Branched chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia 55:321–330
Piraud M, Vianey-Saban C, Petritis K, Elfakir C, Steghens JP, Bouchu D (2005) Ion-pairing reversed-phase liquid chromatography/electrospray ionization mass spectrometric analysis of 76 underivatized amino acids of biological interest: a new tool for the diagnosis of inherited disorders of amino acid metabolism. Rapid Commun Mass Spectrom 19:1587–1602
Waterval WA, Scheijen JL, Ortmans-Ploemen MM, Habets-van der Poel CD, Bierau J (2009) Quantitative UPLC-MS/MS analysis of underivatised amino acids in body fluids is a reliable tool for the diagnosis and follow-up of patients with inborn errors of metabolism. Clin Chim Acta 407:36–42
Acknowledgements
We are grateful to Mr. Abhimanyu Kumar, CEO and Mr. Susheel Sing CTO of General Diagnostics International Pvt Ltd, Mumbai (India), for their institutional funding, continuing interest, encouragement, and administrative support.
Funding
The authors declare that they have no any external agency funding for the present work.
Author information
Authors and Affiliations
Contributions
All authors equally contributed to the study conception and design, material preparation, data collection, and analysis.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Panaskar, S.N., Singh, S.K. Quantification of Amino Acids in Plasma by High-Performance Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS). Chromatographia 86, 567–572 (2023). https://doi.org/10.1007/s10337-023-04262-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-023-04262-3