
Abstract
An environment-friendly method for the fabrication of silica microspheres with tunable pore sizes and narrow particle size distribution was reported through the procedure of twice alkali-thermal reaction, deposition, and calcination. Porous mercaptopropyl-functionalized polysilsesquioxane (Mp-P) microspheres were used as hard templates, the pore size of the Mp-P microspheres can be enlarged by adjusting the reaction pH of the secondary alkali treatment. After modification of the allyl quaternary amine group by the “thiol-ene” click reaction, the mechanical strength of the microspheres can be improved through the electrostatically induced deposition of tetraethyl orthosilicate prepolymer (PES). The effects of the surface charge of template microspheres and the amount of PES deposition on pore structure and mechanical strength of the microspheres were investigated. After calcination, silica microspheres with different pore sizes can be obtained. Propyltrimethoxysilane was bound to the microspheres and used directly as chromatographic stationary phases without classification. The performance of the columns with different pore sizes was evaluated and compared in terms of retention factor, reduced plate height, and resolution for the separation of the protein mixture composed of ribonuclease A, insulin, cytochrome C, and bovine serum albumin. The results showed that the propyl column with a pore size of 18 nm was suitable for the separation of proteins with molecular weights up to 70 kDa, with high column efficiency and resolution.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The promotion of low carbon and environmental protection concept makes people pay more attention to green energy. Developing simple, efficient, and environment-friendly fully porous silica microspheres fabrication technology has become a research hotspot in the field of chromatography [1, 2]. Fully porous materials are comprised of a network of ordered pores, which are associated with a high specific surface area for the packing material and a high peak capacity of the column compare to the core–shell packings. Various methods have been employed to prepare fully porous silica microspheres, among which the sol–gel [3], spray drying [4], polymerization-induced colloid aggregation [5], and template method [6] are widely used. Owing to the high flexibility and versatility, compatibility with a wide range of substances, and less energy consumption, the template method has found extensive applications in the fabrication of silica microspheres. The silica microspheres prepared by the template method can be used directly for chromatographic packing without size classification, which significantly reduces energy consumption [7].
The requirement of relatively large mesopores (> 10 nm) particles for the separation of the biomedical field has been increasing [8]. By using silica particles with large pore sizes, the mass transfer resistance during the chromatographic separation of peptides, proteins, and other biological macromolecules can be greatly reduced [9, 10]. Such large mesopores can be obtained by the soft template method which combines block copolymers and swelling agents. The pores were expanded by introducing auxiliary organic solvents such as trimethylbenzene (TMB) and amines as swelling agents in the hydrothermal treatment. However, the wider pore size distribution or particle fragmentation affects their column efficiency and limits their application at high pressures [11]. Compared with the soft template method, the confining effect of the hard template can effectively control the morphology, particle size, and pore size of the obtained silica particles [12]. The majority of the synthesis methods employ uniform organic or inorganic materials as hard templates, such as polymethylsilsesquioxane (PMSQ) [13], polystyrene [14], polyvinylpyrrolidone [15], calcium carbonate nanoparticles (NPs) [16], and Fe3O4 NPs [17]. Among them, through altering the condensation and alkali thermal reaction conditions, PMSQ microspheres of different particle sizes show good biocompatibility and chemical stability. Huo et al. [18] used porous PMSQ microspheres as hard templates to produce monodisperse porous silica microspheres with 10–12 nm pore sizes. This preparation method is simple, efficient, and energy-efficient, but there is still a lot of work left in terms of aperture control. Chen et al. [12]. prepared monodisperse porous silica with tunable pore size by changing the particle size of the poly (GMA-coEDGMA) microspheres with trimethylamine hydrochloride functionalization as a hard template. By altering the structural properties of the template, mesoporous silica with various pore sizes and morphologies can be obtained.
To get pure inorganic silica beads, the organic moiety of the template needs to be removed, which is often carried out under strong acid or alkali conditions at elevated temperatures. Unfortunately, collapse often accompanies this procedure and leads to the shrinkage of the pore size and poor mechanical strength of the silica beads [19, 20]. Therefore, increasing the mechanical strength of the silica microspheres has been proven to be a viable strategy for maintaining the integrity of the pore structure. To address this problem, He et al. [21]. described a method for producing silica microspheres using ethylenediamine-modified poly (GMA-co-EGDMA) as the template. Tetraethyl orthosilicate (TEOS) was deposited on the template microspheres by the sol–gel process before calcination. The silica microspheres produced by this method showed no shrinkage in particle size which is typically observed in microspheres where the polymer serves as the template. Xia et al. [22]. used tetraethylenepentamine instead of ethylenediamine to produce silica microspheres with pore sizes of 52 nm, and were able to separate proteins well after octadecyl modification. Chen [12] found that trimethylamine-functionalized poly (GMA-co-EGDMA) microspheres can strengthen their interaction with TEOS and the microspheres aggregation was avoided due to TEOS self-nucleation. In our previous work, Bai [23] prepared monodisperse microsized magnetic porous silica microspheres by depositing tetraethyl orthosilicate prepolymer (PES) on the glycidyl methacrylate-ethylene glycol dimethacrylate microspheres for the enrichment of cephalosporins in wastewater. PES has a higher molecular weight than TEOS, which is expected to be more suitable to support the pore structure of the microspheres and enhance their mechanical strength.
In this work, sulfhydryl propyl functional polysiloxane microspheres (Mp-P) were synthesized and used as template microspheres [24,25,26]. A straightforward and environment-friendly strategy to accomplish controllable fabrication of porous silica microspheres with narrow particle size distribution through twice alkali-thermal reactions, electrostatic induced deposition, as well as calcination was reported. The effect of twice alkali-thermal reactions on the particle size and pore structure of Mp-P microspheres was investigated. Through a “thiol-ene” click reaction, the allyl quaternary amine groups were modified to the template microspheres. The deposition mass ratio of the modified template microspheres to PES was optimized and the mechanism of electrostatic induced deposition was elucidated. After calcination, porous silica microspheres with pore sizes of 10, 12, and 18 nm were obtained and subsequently modified with the propyltrimethoxysilane by a vapor deposition method. The prepared columns were evaluated and used for the separation of short-chain peptides and protein macromolecules.
Experimental
Materials
Methyltrimethoxysilane (MTMS), mercaptopropyltrimethoxysilane (MPTMS), tetraethyl orthosilicate (TEOS), and propyltrimethoxysilane were purchased from Wuda Silicone Materials (Wuhan, China). Glycylphenylalanine, boc-L-phenylalanine, fmoc-phenylalanyl-glycine, insulin, cytochrome C, ribonuclease A, and bovine serum albumin were bought from Yuanye Biotechnology (Shanghai, China). Acryloyloxyethyltrimethyl ammonium chloride, azobisisobutyronitrile (AIBN), ammonium hydroxide (25 wt%), sodium hydroxide, ammonium formate, sodium dihydrogenphosphate, potassium dihydrogen phosphate, and other chemicals were obtained from Concord Chemical Reagents (Tianjin, China).
Preparation and Mesopore Expansion of Mercaptopropyl Functionalized Polysilsesquioxane Template Microspheres (Mp-P)
The mercaptopropyl functionalized polysilsesquioxane microspheres (Mp-P) were synthesized according to a previously reported method [24]. They were used as the template microspheres for the fabrication of porous silica beads. To expand the mesopore of the particles, a twice alkali-thermal reaction was performed. In detail, 10 g of the Mp-P template microspheres were dispersed in 50 mL of 80% ethanol containing 0.05 M sodium hydroxide and shaken at 70 °C for 10 h. After the reaction, the microspheres were washed with 95% ethanol and water alternately until the pH of the filtrate was neutral. The microspheres were redispersed in the 80% ethanol containing different concentrations of sodium hydroxide and repeated the above treatment once. After being washed, the products were dried at 90 °C in vacuo. Mercaptopropyl functionalized polysilsesquioxane organic microspheres with different pore sizes can be obtained.
Preparation of Organic–Inorganic Hybrid Microspheres (Mp-N(CH3)3 +-Silica)
Firstly, the above synthesized Mp-p template microspheres with different pore sizes were modified with quaternary amino groups by bonding allyltrimethylammonium chloride in the following process. Mp-p template microspheres (3 g), azobisisobutyronitrile (0.03 g), and allyl trimethyl ammonium chloride (1.95 g) were dispersed in 50 mL of methanol, being shaken at 70 °C for 8 h under a nitrogen atmosphere. The resulting microspheres were isolated by filtration, washed thoroughly with ethanol and distilled water successively, and then dried in vacuo overnight at 90 °C to obtain quaternary amine functionalized polymethylsilsesquioxane microspheres Mp-N(CH3)3+.
Secondly, Mp-N(CH3)3+-silica hybrid microspheres were prepared. Tetraethyl orthosilicate prepolymer (PES) with a certain viscosity was produced according to the previous reporting method [27]. Mp-N(CH3)3+ microspheres (1 g) and different amounts of PES were dispersed in 20 mL absolute ethanol, shaken at room temperature for 3 h, then evaporated to remove the solvent. The products were redispersed in 20 mL 0.25% SDS and ammonia solution, where SDS solution was used as an emulsifier, ammonia increased the degree of cross-linking of PES and regulated the pore structure. The mixture was shaken for 24 h, washed continuously with ethanol and distilled water, and dried at 90 °C in vacuo. The obtain organic–inorganic hybrid microspheres were named Mp-N(CH3)3+-silica.
Preparation and Modification of Silica Microspheres
The above synthesized Mp-N(CH3)3+-silica hybrid microspheres were calcined in a muffle furnace at 600 °C for 6 h to obtain inorganic silica microspheres with different pore sizes. Silica microspheres with pore sizes of 18 and 12 nm were obtained by calcining template beads with a pore size of 18 nm through depositing or not depositing PES, respectively. Silica microspheres with a pore size of 10 nm were obtained by depositing TEOS instead of PES on the template bead with a pore size of 10 nm. After activating the silanol groups by treating them in a 10% hydrochloric acid solution, the obtained microspheres were modified with the propyl group by a vapor deposition method. In detail, 3 g of activated silica microspheres and 1.7 mL of propyltrimethoxysilane were dispersed in 30 mL of dichloromethane and shaken at room temperature for 3 h. After evaporation of the dichloromethane, the particles were transferred to an autoclave, and 1.3 mL of anhydrous triethylamine was added to react at 150 °C for 8 h. The products were washed with dichloromethane, 95% ethanol, and water in turn before being dried at 90 °C in vacuo.
Characterization
A field emission scanning electron microscope (SEM), Nanosem 430, was applied to determine the morphology and structure of the microspheres. The particle size was measured by a particle size analyzer (Beckman Coulter Multisizer 3, Brea, CA, USA). The pore textural properties of the microsphere were detected on the Quantachrome NOVA 3000 surface area analyzer (Boynton Beach, FL, USA). Fourier transform infrared spectroscopy (FT-IR) of the microspheres was analyzed on a Nicolet 6700 with a resolution of 4 cm−1. The carbon and nitrogen concentrations were determined by elemental analysis (Vario EL III, Hanau, Germany). The zeta potential of the microspheres was measured by the Zetasizer Nano ZS (Malvern, UK).
Application for Peptide and Protein Separation
The prepared propyl-silica stationary phases with 10, 12, and 18 nm pore sizes were packed into stainless-steel columns (100 × 4.6 mm) through high-pressure slurry techniques to obtain the silica-10-C3, silica-12-C3, and silica-18-C3 columns, respectively. The chromatographic evaluation was conducted on an Agilent 1100 series HPLC system equipped with a quaternary pump (G1311A), vacuum degasser (G1322A), autosampler (G1367A), column oven (G1316A), variable wavelength UV–vis detector (G1314A), and ChemStation version B 03.01 (Palo Alto, CA, USA).
The mechanical strength of three as-prepared columns was investigated before being used under the chromatographic conditions of methanol as the mobile phase; temperature, 25 °C; flow rate, 0.5–2.5 mL min−1, detection wavelength, 254 nm.
The Van Deemter's (V-D) curves of the above columns were determined using naphthalene as a probe. Mobile phase, methanol–water (80/20, v/v); temperature, 25 °C; flow rate, 0.1–1.5 mL min−1; detection wavelength, 254 nm.
The short-chain peptides sample solution consisted of glycylphenylalanine (2 mg mL−1), boc-L- phenylalanine (5 mg mL−1), and fmoc-phenylalanyl-glycine (7 mg mL−1) were dissolved in 30 mM ammonium formate (pH 6.3). Chromatographic separation conditions, ammonium formate-methanol (pH 6.3, 30 mM, 50:50, v/v); column temperature, 25 °C; flow rate, 1 mL min−1; detection wavelength, 254 nm; sample injection, 20 μL.
The protein sample solution consisted of insulin (5 mg mL−1), ribonuclease A (10 mg mL−1), cytochrome C (3 mg mL−1) and bovine serum albumin (6 mg mL−1) were dissolved in sterilized water. Mobile phase A, phosphate buffer (pH 3; 10 mM) − 0.1% formic acid; mobile phase B, acetonitrile; gradient elution program: 0–30 min, 20–50% acetonitrile; temperature, 25 °C; flow rate, 0.7 mL min−1; detection wavelength, 280 nm; sample injection, 20 μL.
Results and Discussion
Mesopores Expansion of Mp-P Template Microspheres by the Twice Alkali-Thermal Reaction
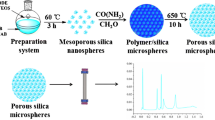
As illustrated in Fig. 1, porous silica microspheres were fabricated using porous mercaptopropyl polysiloxane (Mp-P) microspheres as the innovative hard template, and pore enlarged Mp-N(CH3)3+ microspheres were synthesized via two-step alkali-thermal treatment and “thiol-ene” click chemistry reaction. To maintain the pore structure and increase the mechanical strength of the produced silica microspheres, PES was deposited on the Mp-N(CH3)3+ microspheres to prepare Mp-N(CH3)3+-silica hybrid microsphere. Organic moieties of the hybrid microsphere were removed through calcination, and silica beads can be obtained.

Diagram of silica microspheres preparation
Sodium hydroxide and ammonia were commonly used to regulate the pore structure of Mp-P microspheres under elevated temperatures. According to the literature, microspheres with tunable pore sizes of 5–12 nm could be synthesized under different alkali concentrations [24, 25]. Increasing the alkali concentration helps to obtain microspheres with larger pore sizes. However, the enlargement of the pore size was limited by structural fragmentation or the formation of microsphere gelatinous precipitates under higher sodium hydroxide concentrations. To address this issue, a twice alkali-thermal procedure was developed through a low-concentration primary alkali-thermal reaction followed by a secondary alkali-thermal treatment at a higher concentration to broaden the pore size.
The Mp-P template microspheres were treated at a primary alkali-thermal reaction under an optimized 0.05 M concentration of sodium hydroxide, ensuring the structural integrity of the microspheres and at the same time optimizing the pore size of the particles at approximate 10 nm. After being washed to neutral, the microspheres underwent a secondary alkali-thermal reaction at different sodium hydroxide concentrations of 0–0.7 M. The effects on the particle size and pore structure were investigated, and the results were shown in Table 1 and Fig. 2. It can be seen that the particle size of the Mp-P template gradually decreased from 6.17 to 3.68 μm as the sodium hydroxide concentration increased from 0 to 0.7 M, and the hysteresis loop of the isothermal curve moved to the high-pressure region. According to the BJH pore size distribution, the pore size gradually increased from 9.68 to 32.5 nm and the specific surface area decreased from 438 to 207 m2 g−1.

The pore structure of Mp-P under secondary alkali-thermal with different sodium hydroxide concentrations. a Adsorption–desorption isotherms and b pore size distribution
These results proved the alkali solution could corrode both the surface and the interior of the microspheres. The strong corrosion effect on the outer surface caused the silica nanoparticles to dissolve, resulting in a reduction in particle size. At the same time, more pore channels were formed by the coalescence and stacking of silica nanoparticles. The original convex areas were dissolved under alkali-thermal conditions, resulting in larger pore channels [28]. The twice alkali-thermal reaction, which was considered to be a stepwise expansion method, simultaneously took into account preserving the particle size of the microspheres while achieving an expansion of the pore size. It promoted the polymerization reaction between silanol groups at low sodium hydroxide concentration to improve the alkali resistance of the microspheres, then expanded the pore size through corrosive action at high sodium hydroxide concentration. The pore structure could be precisely regulated by altering the alkali concentration in the secondary alkali thermal treatment.
To obtain template Mp-P microspheres with a diameter of 5 μm, a lower pH value and longer reaction time were necessary during the co-condensation step in the production of the mercaptopropyl functionalized polysilsesquioxane template microspheres. Moreover, a relatively mild secondary alkali thermal reaction at 0.5 M sodium hydroxide concentration was used to synthesize the template microspheres with a 5 μm diameter and a pore size of 18 nm for subsequent studies.
Electrostatic Induced Deposition for Silica Pore Structure Preservation
Through twice alkali-thermal reaction, the pore size of Mp-P microspheres was enlarged, however, after calcination, it was reduced dramatically from 18 to 12 nm. This was ascribed to the collapse of the pore structures when removing the template at elevated temperatures. Therefore, increasing the rigidity and mechanical strength of silica microspheres was a feasible way to maintain the pore structure. According to the literature, tetraethyl orthosilicate (TEOS) or its prepolymer (PES) can be deposited on the porous polymer microspheres to avoid the collapse of the pore structure [12, 19, 21]. Inspired by these works, PES was deposited into the pores of the Mp-P template microspheres before calcination to maintain the pore structure and improve their mechanical strength. To investigate the effect of template surface charge on the deposition, positively charged Mp-N(CH3)3+ microspheres were prepared for comparison by grafting quaternary amine groups onto the surface of Mp-P microspheres through “thiol-ene” click chemistry method. The reaction takes a small amount of methanol and water as solvents, avoids the use of toxic aromatic reagents, has high efficiency, few by-products, and conforms to the principles of less hazardous chemical synthesis in “green chemistry” and the principle of safer solvents and auxiliaries. PES was deposited on both Mp-P and Mp-N(CH3)3+ microspheres at the same mass ratio for comparison. In addition, the effect of PES addition to the Mp-N(CH3)3+ was investigated at a mass ratio of 2:1, 1:1, and 2:3, respectively.
The zeta potential and specific particle sizes of Mp-P, Mp-N(CH3)3+, and Mp-N(CH3)3+-PES hybrid microspheres were listed in Table 2. It could be seen that the PES had a negative charge of − 8.58 mv, while Mp-P microspheres had an almost neutral zeta potential. After modification with quaternary amine groups, the Mp-N(CH3)3+ microspheres showed a positive charge of 25.8 mv, which was expected to deposit more amount of PES. This was confirmed by comparing the particle sizes of Mp-P-silica and Mp-N(CH3)3+-silica prepared under the same deposition ratio at 2:1. Larger particle sizes and more narrow particle size distribution were observed at the Mp-N(CH3)3+-silica hybrid microspheres. This demonstrated the process was controlled by an electrostatic induction deposition mechanism. With the increase of PES addition, the particle size of the Mp-N(CH3)3+-silica hybrid microspheres gradually increased and the particle size distribution further narrowed.
The deposition ratio of PES inside versus outside the template microsphere can be calculated by measuring the particle size and density of the Mp-N(CH3)3+-silica hybrid microspheres [23]. As shown in Table S1, it could be inferred that PES was deposited on the surface and interior of the template microspheres at an approximate ratio of 57:43 with an optimized Mp-N(CH3)3+: PES deposition ratio of 2:1. As the amount of PES increased, the proportion distributed outside the pore increased. To elucidate the effect of deposition on pore structure, nitrogen adsorption–desorption detection was performed on the Mp-P and silica microspheres obtained by calcination of different deposition ratios. As shown in Fig. 3a, the type IV adsorption–desorption isotherms were observed, which proved the presence of the mesoporous structures. Based on the pore size distribution curve shown in Fig. 3b, a dramatical decrease in the pore size was observed for the silica beads produced by direct calcination without disposition of any PES (PES = 0). The pore size collapsed from 18 to 12 nm due to the lack of structural rigidity. In contrast, the silica microspheres obtained by calcination with Mp-N(CH3)3+: PES = 2:1 maintained the pore size of 18 nm to the greatest extent. As the deposited PES further increased, a large number of 4–5 nm micropores were observed. At the same time, PES would be more likely to be deposited on the surface of the template microsphere, resulting in an increase in particle size.

The pore structure of silica microspheres under the different templates or PES additions. a Adsorption–desorption isotherms and b pore size distribution
The surface of template microspheres modified with positively charged quaternary amino groups had an electron-donating effect and thus easily interacted electrostatically with the negatively charged nano-particles or oligomers formed by PES hydrolysis. This procedure induced more nano-particles to be deposited into the template microsphere and avoided the self-nucleation of PES. When the mass ratio was up to 2:3, the pore structure channels of the Mp-N(CH3)3+ microspheres were filled with excessive silica nanoparticles produced by PES hydrolysis, resulting in a significant increase in micropores after calcination. These micropores might be interstitial pores generated by the secondary spheroid formation of PES at high concentrations. In addition, the particle size of the resulting silica beads after calcination was reduced by approximately 20%, which was reasonable for silica beads prepared by the template method. The detailed parameters of particle size and pore structure parameters are shown in Table 3. Through the electrostatically induced deposition process, the pore walls became thicker and the mechanical strength of the silica was expected to be increased.
Characterization
The morphology and the particle size distribution of Mp-N(CH3)3+, Mp-N(CH3)3+-silica hybrid microspheres, and silica beads were determined by field emission scanning electron microscopy and a particle size analyzer. As shown in Fig. 4 and Table S2, all these microspheres were in spheric and uniformed shape. The presence of some flake structures may come from the corrosive effect of a strong alkali solution, which can be removed by subsequent washing procedures. After deposition by the PES, the Mp-N(CH3)3+-silica hybrid microspheres showed better spherical regularity and a narrow particle size distribution. Through calcination, the particle size of the silica microspheres reduced from 5.68 to 4.58 μm. This was attributed to the removal of organic moieties from the hybrid microspheres and also the compaction of the silica moieties during the calcination.

SEM micrograph and particle size distribution of the a Mp-N(CH3)3+, b Mp-N(CH3)3+-silica, c silica
The functional groups in Mp-P, pore expanded Mp-P, Mp-N(CH3)3+, Mp-N(CH3)3+-silica, and silica microspheres were confirmed by the FT-IR and their spectra were shown in Fig. S1. On the Mp-P microspheres, the region of 3750–3000 cm−1 was the absorption of the O–H. 1132 cm−1 and 1032 cm−1 were the characteristic absorption peaks of the Si–O-Si bond of polysesquioxane. The signal at 840 cm−1 was the bending vibration peak of Si–C. The Si–O-Si signal on the pore expanded Mp-P microspheres at 1132 and 1032 cm−1 was stronger than that of Mp-P, indicating that the twice alkali thermal reaction increased the degree of crosslinking of the silanol. The absorption peak of Mp-N(CH3)3+ at 1620–1570 cm−1 was the characteristic absorption of C-N, proving that the quaternary amine group had been successfully bonded. The enhanced Si–O-Si characteristic absorption peak of Mp-N(CH3)3+-silica at around 1100 cm−1 was caused by the deposition reaction. After calcination, the signal of O–H absorption on the silica microspheres was enhanced, both the C-H stretching vibration and C-N characteristic absorption peak disappeared, indicating the successful removal of carbon and nitrogen elements. This conclusion was consistent with the results of the elemental analysis determination of less than 1% of the carbon and nitrogen contents shown in Table S2.
The pore structures of the synthesized silica microspheres with 10, 12, and 18 nm pore sizes were shown in Fig. S2 and the second and fourth curves of Fig. 3, respectively. It can be seen that the pore size distribution was narrow and there were no mesopores of 4–5 nm. After being modified with propyltrimethoxysilane through the vapor deposition method, a particle size analyzer and elemental analysis measurements were performed and shown in Table S3. As can be seen, they showed a particle size of approximately five microns and a narrow particle size distribution with the value of D90/D10 in the range of 1.2–1.3, even better than the product obtained by the emulsion polymerization method after classification. The average bonding density of the propyl groups was calculated according to the equation developed by Berendsen and de Galan [29] with the value of 1.52, 1.39, and 2.06 µmol m−2 on silica-10-C3, silica-12-C3, and silica-18-C3, respectively.
After packing the above-prepared silica stationary phase into the steel tube, the mechanical strength of the three columns was measured by plotting the pressure–velocity curves. As shown in Fig. S3, the correlation coefficient of the fitted curve (R2) of all the columns was greater than 0.99 with good linearity, indicating the presence of high mechanical strength and regular column bed structure [30]. The higher column pressure compared to the other two columns is attributed to the smaller particle size of 4.39 μm in the silica-18-C3 column.
The V-D equation was used to analyze the efficiency and mass transfer characteristics of different columns. As shown in Fig. S4, at lower flow rates, the silica-12-C3 and silica-18-C3 columns showed higher column efficiencies. When the flow rate was higher, the silica-10-C3 column exhibited a higher column efficiency. The V-D equation parameters of silica-10-C3, silica-12-C3, and silica-18-C3 columns are listed in Tables S3. The minimum reduced plate heights of these three columns were 2.88, 2.507, and 2.506, respectively, reflecting a different column bed structure. The A term was the smallest for the silica-18-C3 column at 0.975, which coincides with its uniform particle distribution D90/D10 value of 1.23. The C term corresponding to the mass transfer resistance of the silica-18-C3 was higher than the other two columns which may relate to its irregular pore structure. Based on the adsorption–desorption isotherms of Fig. 3, the hysteresis loop type of 18 nm silica packing is biased towards the H2 type. The presence of ink-bottle-shaped pores and some micropores results in the stagnation of analytes thus increasing the mass transfer resistance.
Separation of Peptides and Proteins
It is well known that the pore size of chromatographic packing plays a significant effect on the performance of chromatographic separation [31]. The pore size determines whether target molecules can enter and exit the pore channel. A relatively large, well-regulated pore structure can reduce the mass transfer resistance and improve column efficiency. The performance of three stationary phases including silica-10-C3 (5.68 μm, 10 nm), silica-12-C3 (5.17 μm, 12 nm), and silica-18-C3 (4.39 μm, 18 nm) was explored by three short peptides and four proteins with different molecular weight. The effect of the pore size of the stationary phase was elucidated through these evaluations.
The chromatographic conditions for short peptides and proteins were referred to in the work of previous investigators [32]. All three peptides including glycylphenylalanine, boc-L-phenylalanine, and fmoc-phenylalanyl-glycine were dissolved in the mobile phase, reducing baseline perturbations in the HPLC system. It can be seen in Fig. 5a, that baseline separation of the short peptides was achieved within 16 min on all three columns. As shown in Table S4, the retention factors, theoretical plate number and separation resolution for the peptides were raised as the pore size of the columns increased, and the reduced plate height gradually decreased. Lower reduced plate height and higher resolution were observed on the silica-18-C3 column for the separation of fmoc-phenylalanyl-glycine with a molecular weight up to 444 Da. This is attributed to the smaller particle size and narrower particle size distribution, which correlated with the value of the smallest A value in its V-D parameters.

Separation of peptides and proteins. a Samples: (1) glycylphenylalanine; (2) boc-L-phenylalanine; (3) fmoc-phenylalanyl-glycine. Mobile phase, methanol-ammonium formate buffer (pH 6.3; 30 mM, 30/70, v/v); flow rate: 1.0 mL min−1; temperature: 25 °C; wavelength: 254 nm. b Samples: (1) ribonuclease A; (2) insulin; (3) cytochrome C; (4) bovine serum albumin. Mobile phase, A: PBS (10 mM) − 0.1% formic acid, B: acetonitrile; gradient: 20–50% acetonitrile in 0–30 min; flow rate, 0.7 mL min−1; temperature: 25 °C; wavelength: 280 nm
Ribonuclease A, insulin, cytochrome C, and bovine serum albumin were used as model proteins to evaluate the performance of these three columns and the results were shown in Fig. 5b. The addition of 0.1% v/v formic acid to the mobile phase acted as an ion-pair reagent to interact with positively charged and polar groups of the protein and reduce non-specific adsorption [33]. In addition, gradient elution was chosen to shorten the analysis time and improve the peak shape of the proteins with different hydrophobicity. After optimization of mobile phase pH and gradient conditions, the separation resolution was significantly improved.
Hydrophobic, ion exchange, hydrogen bonding, and hydrophilic interactions are present in the separation of proteins. For the as-prepared propyltrimethoxysilane modified chromatographic columns, hydrophobic forces played a dominant role in the separation process. As shown in Fig. 5b, four proteins reached baseline separation on three columns within 25 min in the order of ribonuclease A, insulin, cytochrome C, and bovine serum albumin. The molecular weight, pI values, and gyration diameter [12] of the proteins were shown in Table S5. Their separation parameters were listed in Table S6. As the pore size of the three columns increased, the retention factor, theoretical plate number, and separation resolution significantly improved and the reduced plate height gradually decreased. It was observed on the silica-10-C3 column that the small pore size resulted in a broadening of the chromatographic peak and a decrease in column efficiency. The reduced plate height of 0.93 on the silica-12-C3 column for the separation of bovine serum albumin was higher than that of 0.51 on the silica-10-C3. This might be ascribed to the relatively high mass transfer resistance of the silica-12-C3 column for the large molecular weight analytes. Among them, the silica-18-C3 column exhibited sharper peaks, higher column efficiency as well as better resolution. This demonstrated that the pore size of 18 nm was large enough for proteins with a diameter of fewer than 3 nm such as insulin, ribonuclease A and cytochrome C to access pores without restricted diffusion [34]. Compared to the similar work reported by Chen et al. [12], the half-peak widths of the silica-18-C3 we prepared were 0.53 and 0.18 for the separation of ribonuclease A and cytochrome C, achieving comparable column efficiency. In addition, bovine serum albumin with a diameter of about 5 nm, also showed good separation performance on the silica-18-C3 column, with a theoretical plate number of 10.46 × 104 per meter. This was similar to the column performance of a C18-modified, 22.3 nm pore size core–shell dioxide column prepared by Qu et al. [35]. Furthermore, as shown in Fig. S4, the silica-18-C3 column exhibited excellent reproducibility for the separation of proteins.
Conclusion
The hard template method has proven to be an environmentally friendly method of fabricating silica microspheres with a narrow particle size distribution and variable pore sizes, which can be directly used as chromatographic packing without multi-stage sieving and avoids the high energy consumption, in line with the energy-efficient design principle of green chemistry. Porous mercaptopropyl-functionalized polysilsesquioxane mesoporous microspheres (Mp-P) were used as hard templates, and a series of silica with narrow particle size distribution and adjustable pore sizes were prepared. Through a twice alkali-thermal process, Mp-P microspheres with pore sizes ranging from 9 to 33 nm were produced. After introducing quaternary ammonium groups to the template Mp-P microspheres, electrostatic induced deposition and calcination procedures helped to create silica with large pore size and high mechanical strength. Under the optimized amount of tetraethoxysilane prepolymer (PES) deposition, the synthesized silica microspheres showed good mechanical strength and large pore sizes. After modification with propyltrimethoxysilane, the performance of the columns was evaluated. It can be seen that the retention factor, theoretical plate number, and separation resolution gradually increased with the increase of the pore size. The results illustrated that the silica-18-C3 column allowed the free diffusion of proteins with molecular weights up to 70 kDa with high column efficiency. This method is expected to be used for the preparation of silica microspheres with even larger pore sizes, and work is underway.
References
Qiang TT, Zhu RT (2022) Bio-templated synthesis of porous silica nano adsorbents to wastewater treatment inspired by a circular economy. Sci Total Environ 819:152929. https://doi.org/10.1016/j.scitotenv.2022.152929
Zhao Y, Wang J, Yang Y, Fu Q, Ke Y (2022) Pseudomorphic synthesis of bimodal porous silica microspheres for size-exclusion chromatography of small molecules. J Chromatogr A 1664:462757. https://doi.org/10.1016/j.chroma.2021.462757
Vorotyntsev AV, Markov AN, Kapinos AA, Petukhov AN, Pryakhina VI, Nyuchev AV, Atlaskina ME, Andronova AA, Markova EA, Vorotyntsev VM (2020) Synthesis and comparative characterization of functionalized nanoporous silica obtained from tetrachloro- and tetraethoxysilane by a sol-gel method. Phosphorus Sulfur 196:176–188. https://doi.org/10.1080/10426507.2020.1825433
Zhang HW, Li ML, Li JM, Agrawal A, Hui HW, Liu DM (2022) Superiority of mesoporous silica-based amorphous formulations over spray-dried solid dispersions. Pharmaceutics 14:428–443. https://doi.org/10.3390/pharmaceutics14020428
Yang XH, Wan GP, Ma SJ, Xia HJ, Wang J, Liu JW, Liu YN, Chen G, Bai Q (2020) Synthesis and optimization of SiO2@SiO2 core-shell microspheres by an improved polymerization-induced colloid aggregation method for fast separation of small solutes and proteins. Talanta 207:120310. https://doi.org/10.1016/j.talanta.2019.120310
Khoeini M, Najafi A, Rastegar H, Amani M (2019) Improvement of hollow mesoporous silica nanoparticles synthesis by hard-templating method via CTAB surfactant. Ceram Int 45:12700–12707. https://doi.org/10.1016/j.ceramint.2019.03.125
Shi Y, Wang JZ, Yamamoto E, Osada M (2020) Hard-template synthesis of hollow mesoporous silica nanoplates using layered double hydroxide. Chem Lett 49:1078–1080. https://doi.org/10.1246/cl.200387
Muramoto N, Sugiyama T, Matsuno T, Wada H, Kuroda K, Shimojima A (2020) Preparation of periodic mesoporous organosilica with large mesopores using silica colloidal crystals as templates. Nanoscale 12:21155–21164. https://doi.org/10.1039/d0nr03837g
Wu YL, Sun XT, Wang HY, Shen JW, Ke YX (2022) Pore size control of monodisperse mesoporous silica particles with alkyl lmidazole Ionic liquid templates for high performance liquid chromatography applications. Colloid Surf A 637:128200. https://doi.org/10.1016/j.colsurfa.2021.128200
Chen X, Zhang S, Hou D, Duan H, Deng B, Zeng Z, Liu B, Sun L, Song R, Du J, Gao P, Peng H, Liu Z, Wang L (2021) Tunable pore size from sub-nanometer to a few nanometers in large-area graphene nanoporous atomically thin membranes. ACS Appl Mater Inter 25:29926–29935. https://doi.org/10.1021/acsami.1c06243
Ahmed A, Myers P, Zhang HF (2014) Synthesis of nanospheres-on-microsphere silica with tunable shell morphology and mesoporosity for improved HPLC. Langmuir 30:12190–12199. https://doi.org/10.1021/la503015x
Chen JW, Zhu LL, Ren LB, Teng C, Wang Y, Jiang BW, He J (2018) Fabrication of monodisperse porous silica microspheres with a tunable particle size and pore size for protein separation. ACS Appl Bio Mater 1:604–612. https://doi.org/10.1021/acsabm.8b00088
Meng GD, Li YM, Wang ZD, Pan C, Gao WW, Cheng YH (2021) Preparation and characterization of narrow size distribution PMSQ microspheres for high-frequency electronic packaging. Materials 14:4233. https://doi.org/10.3390/ma14154233
Akhondi M, Jamalizadeh E (2021) Preparation of cubic and spherical hollow silica structures by polystyrene-poly diallyldimethylammonium chloride and polystyrene-poly ethyleneimine hard templates. Ceram Int 47:851–857. https://doi.org/10.1016/j.ceramint.2020.08.197
Zhu YF, Shi JL, Chen HR, Shen WH, Dong XP (2005) A facile method to synthesize novel hollow mesoporous silica spheres and advanced storage property. Micropor Mesopor Mat 84:218–222. https://doi.org/10.1016/j.micromeso.2005.05.001
Le Y, Chen JF, Wang WC (2004) Study on the silica hollow spheres by experiment and molecular simulation. Appl Surf Sci 230:319–326. https://doi.org/10.1016/j.apsusc.2004.02.042
Yang J, Lee J, Kang J, Lee K, Suh JS, Yoon HG, Huh YM, Haam S (2008) Hollow silica nanocontainers as drug delivery vehicles. Langmuir 24:3417–3421. https://doi.org/10.1021/la701688t
Huo ZX, Wan QH, Chen L (2020) Energy-efficient and environment-friendly method to prepare monodispersed silica stationary phases for simultaneous separation of compound drugs. J Chromatogr A 1618:460866. https://doi.org/10.1016/j.chroma.2020.460866
Bianconi LP, Taviot-Gueho C, Constantino VRL, Bizeto MA (2022) Evaluation of the structural integrity of layered double hydroxides and mesoporous silica during the preparation of heterostructures. J Braz Chem Soc 00:1–8. https://doi.org/10.21577/0103-5053.20220041
Savic S, Vojisavljevic K, Pocuca-Nesic M, Zivojevic K, Mladenovic M, Knezevic N (2018) Hard template synthesis of nanomaterials based on mesoporous silica. Metall Mater Eng 24:225–241. https://doi.org/10.30544/400
He J, Yang C, Xiong X, Jiang B (2012) Preparation and characterization of monodisperse porous silica microspheres with controllable morphology and structure. J Polym Sci Pol Chem 50:2889–2897. https://doi.org/10.1002/pola.26066
Xia HJ, Wan GP, Zhao JL, Liu JW, Bai Q (2016) Preparation and characterization of monodisperse large-porous silica microspheres as the matrix for protein separation. J Chromatogr A 1471:138–144. https://doi.org/10.1016/j.chroma.2016.10.025
Bai RT, Chang CQ, Chen L (2020) Preparation of highly responsive monodisperse magnetic porous silica microspheres for the enrichment of cephalosporins in wastewater. J Sep Sci 43:3765–3774. https://doi.org/10.1002/jssc.202000589
Shi JJ, Zhang LX, Huo ZX, Chen L (2021) High stability amino-derived reversed-phase/anion-exchange mixed-mode phase based on polysilsesquioxane microspheres for simultaneous separation of compound drugs. J Pharmaceut Biomed 203:114187. https://doi.org/10.1016/j.jpba.2021.114187
Li JL, Huo ZX, Chen L, Wan QH (2017) Mercaptopropyl functionalized polymethylsilsesquioxane microspheres prepared by co-condensation method as organosilica-based chromatographic packings. Chromatographia 80:1287–1297. https://doi.org/10.1007/s10337-017-3349-4
Huo ZX, Chen L (2020) Base-deactivated and alkaline-resistant chromatographic stationary phase based on functionalized polymethylsilsesquioxane microspheres. J Sep Sci 43:389–397. https://doi.org/10.1002/jssc.201900634
Zhou XL, Wan QH (2015) Separation and identification of oligomeric ethyl silicates by liquid chromatography with electrospray ionization mass spectrometry. J Sep Sci 38:1484–1490. https://doi.org/10.1002/jssc.201401184
Huo ZX, Wan QH, Chen L (2018) Synthesis and evaluation of porous polymethylsilsesquioxane microspheres as low silanol activity chromatographic stationary phase for basic compound separation. J Chromatogr A 1553:90–100. https://doi.org/10.1016/j.chroma.2018.04.024
Soliven A, Dennis GR, Guiochon G, Hilder EF, Haddad PR, Shalliker RA (2010) Cyano bonded silica monolith-development of an in situ modification method for analytical scale columns. J Chromatogr A 1217:6085–6091. https://doi.org/10.1016/j.chroma.2010.07.052
Yildirim D, Gokcal B, Buber E, Kip C, Demir MC, Tuncel A (2021) A new nanozyme with peroxidase-like activity for simultaneous phosphoprotein isolation and detection based on metal oxide affinity chromatography: monodisperse-porous cerium oxide microspheres. Chem Eng J 403:126357. https://doi.org/10.1016/j.cej.2020.126357
Rusli H, Putri RM, Alni A (2022) Recent developments of liquid chromatography stationary phases for compound separation: from proteins to small organic compounds. Molecules 27:907. https://doi.org/10.3390/molecules27030907
Wang JZ, Wang JS, Ning XH, Liu J, Xia H, Wan G, Bai Q (2021) pH-dependent selective separation of acidic and basic proteins using quaternary ammoniation functionalized cysteine-zwitterionic stationary phase with RPLC/IEC mixed-mode chromatography. Talanta 225:122084. https://doi.org/10.1016/j.talanta.2021.122084
Zhao LS, Li SS, Wang WH, Wang YH, Du KF (2021) Preparation and characterization of highly porous cellulose-agarose composite chromatographic microspheres for enhanced selective separation of histidine-rich proteins. J Chromatogr A 1637:461831. https://doi.org/10.1016/j.chroma.2020.461831
Qu Q, Si Y, Xuan H, Zhang K, Chen X, Ding Y, Feng S, Yu HQ, Abdullah MA, Alamry KA (2018) Dendritic core-shell silica spheres with large pore size for separation of biomolecules. J Chromatogr A 1540:31–37. https://doi.org/10.1016/j.chroma.2018.02.002
Qu Q, Si Y, Xuan H, Zhang K, Chen X, Ding Y, Feng S, Yu H-Q (2018) Synthesis of core-shell silica spheres with tunable pore diameters for HPLC. Mater Lett 211:40–42. https://doi.org/10.1016/j.matlet.2017.09.087
Acknowledgements
We thank Dr. Xing-Hua Jin and Yan Gao from the Analytical Center of the School of Pharmaceutical Science & Technology for their assistance in the measure of elemental analysis and zeta potential.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
YJ: investigation, analysis, data curation and writing the original draft. XG: silica microsphere synthesis conditions optimization. CQ: basic tests on columns. LC: supervision, resources, project administration, review and edit of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jing, Y., Guo, X., Qi, C. et al. Fabrication of Silica Microspheres for HPLC Packing with Narrow Particle Size Distribution and Different Pore Sizes by Hard Template Method for Protein Separation. Chromatographia 85, 985–995 (2022). https://doi.org/10.1007/s10337-022-04200-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-022-04200-9