
Abstract
Pharmaceutical regulators are worried about medication quality and stability since drug degradation may result in harmful chemicals. Erlotinib (ERL) is a tyrosine kinase inhibitor associated with the epidermal growth factor receptor (EGFR) containing susceptible functional groups such as quinazoline and amine ketone, methoxy, and ethoxy leads to a reduction in pharmaceutical quality. According to the ICH-Q1A (R2) guideline, the goal of ERL stability studies is to establish its susceptibility to degradation under various environmental conditions. A novel isocratic stability–indicating liquid chromatography method has been developed using systemic quality by design (QbD) approach. The QbD strategy includes screening and optimization as phases. Placket Burman was used for primary parameters screening, and critical factors were optimized with response surface design. The prepared degradation samples (acid, base, neutral hydrolysis, oxidative, photolytic, and thermal) were separated using a Shimadzu GIST C18 column (250 mm × 4.6 mm, 5 µm) with 15 mM ammonium formate: ACN (58:42% v/v) as mobile phase, 0.9 mL/min flow rate, and 246 nm wavelength, which was found to be LC–MS compatible. A total of six degradation products (DPs) were identified with the optimized chromatography method. The drug was sensitive toward acidic and basic hydrolysis, but it remained stable under neutral, oxidative, thermal, and photolytic stress conditions. The optimized method was sensitive, specific, and robust, with linearity ranging from 10 to 35 µg/mL, with a correlation coefficient (R2 = 0.9997). The analytical method greenness score was calculated and observed that the developed method is green.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many drug withdrawals have lately happened due to an impurity in medicines that renders them ineffective. Consequently, the separation between each potential degradation peak and the drug peak must be determined [1]. Moreover, drug recalls by the FDA also raise concerns about pharmaceutical product quality and safety. Stability testing is now a crucial component of the pharmaceutical industry’s process for evaluating the quality of a pharmacological substance or a pharmaceutical product [2]. All drug compounds and drug products deteriorate over time due presence of light, pH, moisture, heat, and other conditions [3]. The International Conference on Harmonization (ICH) guideline has harmonized protocols by considering these problems (safety, efficacy and quality). A stability-indicating test is required to analyze chemical, physical or microbiological factors' impact on final formulation efficiency and integrity [4].
Erlotinib hydrochloride (ERL) is a targeted anticancer drug that inhibits intracellular tyrosine kinase activity associated with the epidermal growth factor receptor (EGFR). ERL is used to treat non-small cell lung cancer (NSCLC) and pancreatic cancer [5, 6]. Lung cancer is the primary cause of cancer-related mortality globally [7]. More than half of patients with NSCLC are too advanced to be operated on, and typical first-line chemotherapy based on platinum drugs improves survival only marginally [8]. The selected drug is water-insoluble and essentially insoluble in acetonitrile and acetone [9]. Chemically, ERL is an N-(3-ethynylphenyl)-6, 7-bis (2-methoxyethoxy) quinazolin-4-amine having a pKa value of 5.42 (Fig. 1). The functional groups quinazoline, amine, ketone, methoxy, and ethoxy in the ERL structure are more susceptible to degradation when exposed to various environmental conditions. As a result, using proper methods, it is critical to explore in-depth the factors that influence the formation, isolation, and qualification of degradation products. This type of in-depth research can aid in determining the expiry date, packaging conditions, retest period, and storage conditions for ELR during handling [10].

Structure of Erlotinib Hydrochloride
Using a QbD approach to develop a stability-indicating method API is considered required. The quality by Design (QbD) approach has gained the pharmaceutical industry’s attention and regulatory authorities [11]. ICH has defined it as “a systematic approach to development that begins with predefined objectives and demonstrates product and process understanding and process control, based on scientific evidence and quality risk management” [12,13,14]. The scientific knowledge acquired can be utilized to decide method controller settings and manage risks that have been identified [15, 16].
Green analytical chemistry also gains scientists' attention because it makes analytical techniques less harmful to the environment, more human-friendly, and cost reduction [17, 18]. The effect and role of green methods are rapidly expanding across a broad spectrum of analytical procedures, making green method development vital for analytical research [19]. Green chemistry specialists use or invent efficient, low-waste assessment procedures that use chemicals safe for humans and the environment. Analytical chemists play an essential role in establishing the efficacy and safety of new and existing methods. Green analytical chemistry practices include analyzing the smallest amount of samples with the smallest sample size. The green approach saves method costs by eliminating waste and lowering the required solvent. The volume and toxicity of reagents, produced waste, energy demands, the number of procedures, downsizing, and automation are only a few of the numerous parameters taken into account when determining the greenness of the analytical process. The use of greenness assessment standards necessitates specific tools [18, 20, 21].
The drug literature review revealed that few articles had been published on ERL stability [14, 22,23,24,25]. The ERL monograph is given in Indian pharmacopeia without data on impurities [26]. The related substance method provided in IP has mentioned the use of phosphate buffer with gradient programming. The author [25] developed a SIAM for ERL, but the approach appears to be less suited for resolving degradation peaks under different degradation conditions due to the short column length. Lastly, the report published by Mahajan et al. [23] had separated and identified two degradation products using the developed ERL stability method. Still, the technique was less efficient in separating all possible degradation products. The author [14] developed and validated a chromatographic technique based on the QbD method, which was adequate for spiking impurities separation. At the same time, no peak was observed during forced degradation sample analysis. Rao et al. [24] developed an HPLC technique to quantify impurities in ERL. The author has used monosodium phosphate, which is incompatible with LC–MS/MS studies.
Moreover, the proposed technique lacks method greenness since the organic phase was 80%. The recent report published by Latha [22] had developed an RP-HPLC method for quantifying ERL using potassium dihydrogen orthophosphate and acetonitrile (70:30; v/v) as the mobile phase, but its mass is incompatible. All the published work and pharmacopeia monograph have limitations in method efficiency, effective degradation products separation, and MS compatible green method. As a result, the purpose of this work was to use the QbD technique to create unique and validated stability-indicating analytical methods (SIAM). The QbD technique will meet stability method criteria such as suitable peak resolution, robustness, selectivity, specificity, and accuracy, which will benefit industrialists throughout the product approval process and monitoring drug quality. By considering the multivariate approach and green chemistry as an emerging trend in the analytical field [16, 27,28,29], the author aimed to develop a new isocratic, green HPLC technique using a QbD approach to separate all degradants according to ICH standards.
Experimental
Chemicals and Reagents
Erlotinib Hydrochloride (Purity 99.9%) was received as a gift sample from MSN Laboratories Private Limited, Sangareddy, Telangana, India, with the certificate of analysis. The HPLC grade methanol (MeOH) and acetonitrile (ACN) were obtained from SD Fine Chemicals (Mumbai, India). HPLC grade water was prepared by filtrating collected Milli-Q water from Millipore Milli-Q- Plus system (Millipore, Milford, MA, USA). Ammonium formate (NH4HCO2), formic acid (HCOOH), Hydrochloric acid (HCl), sodium hydroxide (NaOH), and hydrogen peroxides (H2O2) were used in an analytical grade from Research-Lab Fine Industries, Mumbai.
Equipment and Instruments
The Shimadzu LC 2010 CHT High-Performance Liquid Chromatography equipped with an autosampler and Photodiode array (PDA) detector having Lab Solutions software was used for chromatographic data acquisition and analysis. The buffer pH was monitored by the Lab India pH meter. Analytical balance (Presissa XB220, India) was used for sample weighing compounds.
The high precision water bath having a temperature controller procured from Meta-lab Ltd (Mumbai, India) was used for the degradation studies. Fourier transform infrared spectrophotometer (FTIR-RX1) was used to identify the functional group. The 0.45 µm membrane filters were used to filter the mobile phase. The Ultrasonic bath (L-45) from Electrosonic industries (Mumbai, India) was used to sonicate the HPLC mobile phase. Thermal degradation was carried out using a hot air oven from Meta-Lab Ltd (Mumbai, India). The State-Ease Design-Expert software version 13.0® license version optimized the method.
Solution Preparation
Preparation of Standard Solution
10 mg of ERL were accurately weighed, dissolved in methanol, and diluted with ACN to 10 mL (1000 µg/mL).
Preparation of Working Solution
1 mL was pipetted out from the stock solution and transferred into a 10 mL volumetric flask, where it was diluted up to 10 mL with the appropriate stressor (100 µg/mL). The drug was studied in all the degradation conditions hydrolysis (acid, alkaline, neutral), oxidative, thermal, and photolytic. Acid hydrolysis was performed using hydrochloric acid, alkaline hydrolysis with sodium hydroxide, neutral hydrolysis with water, oxidative hydrolysis with hydrogen peroxide, and thermal and photolytic degradation with acetonitrile (ACN).
Solution Preparation for Degradation Study
Weighing the ERL 10 mg and transferring it to a volumetric flask, 2 mL methanol was added to the flask to dissolve the powder, followed by volume building up to 10 mL with the relevant stressors (1000 µg/mL). Then, at a predetermined interval, 1 mL of the solution was collected and diluted to 10 mL with ACN: Water (50:50, v/v) to produce a working solution with a concentration of 100 µg/mL. At 60 °C, 70 °C, and 80 °C, hydrolytic degradation (acid, alkaline, and neutral) was performed. 1% H2O2, 6% H2O2, and 10% H2O2 on oxidative degradation were studied. Thermal (dry heat) degradation was obtained by keeping solid samples in the oven for 14 h at 80 °C and 60 °C. Photolytic degradation was achieved by exposing solid-state and solution samples (ACN: Water, 50:50, v/v) to light. The acid and alkaline degradation samples were simultaneously neutralized after the experiment to reduce the chances of secondary degradation.
Implementation of Quality by Design Approach for SIAM Method Development
The systemic quality by design approach follows primary parameter selection, secondary parameter screening, and method optimization. Quality by design used for developing analytical methods is always referred to as AQbD (analytical quality by design) [30, 31]. The AQbD approach begins with the identification of an analytical target profile (ATP), critical method attributes (CMAs), and critical method parameters (CMPs), as shown in Table S1. The drug ERL (100 µg/mL) was studied for all degradation conditions. Preliminary ERL degradation results concluded that the drug is sensitive toward acid hydrolysis, which led to more degradation peaks observed in the acid hydrolytic condition. Hence, the acid hydrolysis sample of ERL 100 (µg/mL) was used for the screening and optimization trials. The stability-indicating method desired to resolve the drug peak from all degradation peaks; hence, the resolution between peaks was considered the response variable in screening and optimization designs. The selected factors need to be assessed with risk identification and ranking to identify critical risk factors that affect the desired method criteria as per ICH Q9 [11, 32].
Risk Assessment
Risk assessment results reported critical method parameters (CMPs), which are high-risk factors that significantly influence the CAAs [33]. The risk assessment process identified potential variables influencing method performance and their causes. Method parameters such as sample preparation method, reaction time, solvent preparation, and instrumental settings such as chromatographic mode, mobile phase ratio, flow rate, injection volume, and so on could all have a role. Based on their criticality and influence on the method CAAs, high-risk method factors were chosen and subject to additional investigation employing a suitable screening and experimental optimization strategy.
Screening Using Plackett–Burman Design
The defined quality target product profiles (QTPPs) and critical quality characteristics (CQAs) were used to screen critical parameters, followed by the implementation of the ICH Q9 quality risk management tool [31]. The Ishikawa (Fishbone) diagram and risk assessment tool was used to select eleven significant parameters that required secondary screening using the Plackett–Burman design (design expert 13) [34]. As aforementioned, the resolution is a crucial response variable between all peaks; hence studied resolution as the response variable. The design results were used to construct half-normal plots and Pareto charts. [34]. Pareto charts were used to illustrate the effect of the independent factor on the required responses. The key factors were then identified and applied to the Response surface design for the optimization method.
Response Surface Optimization
After the primary screening, a factor greatly influenced the response variable (peak resolution) was detected, which prompted further secondary parameter investigation [35, 36]. The secondary parameter was studied with response surface design (RSD). The response surface design was chosen because it permits examining the effects of various factors and their interactions on one or more response variables. The design suggested 30 trials, which were carried out to analyze resolution as the response variable. Design expert v13 was used for all statistical calculations, model derivation, ANOVA results, polynomial equations derivation, desirability plot, contour plot derivation, p-value and F-value calculation. Lastly, a control strategy was designed that summaries a set of controls required based on the current product and process expertise performance. An optimized chromatographic condition was applied to check the drug's degradation under all conditions. The factors' primary interaction and quadratic effects on the required response variables were estimated using a four-factor, five-level Response surface design to optimize chromatographic parameters [37], as shown in Table 4.
Method Validation
The suggested stability-indicating method was validated in compliance as per ICH guideline Q2 (R1). The developed method system suitability variables (peak area, asymmetry, theoretical plate, retention time) were assessed by calculating five standards replicate % RSD data. The acid degradation solution was used for the linearity and prepared serial dilutions in a concentration range of 10–50 µg/mL, and each concentration was studied six times (n = 6). An analytical procedure’s precision refers to how closely a set of measurements is obtained from repeated samplings. The precision (Interday and Intraday) was studied in triplicates three times or days at three concentrations (10, 20, and 30 µg/mL). The method accuracy was tested with three distinct concentrations (level 80, level 100, and level 120% of the drug; 18, 20, and 22 µg/mL, respectively), which spiked with a known amount of substance. The percentage recovery was calculated for each prepared sample solution. The response standard deviation and slope value are used to calculate the limit of detection (LOD) and limit of quantification (LOQ) [38]. The found theoretical values sample was prepared and injected into the system and confirmed the limit of detection and quantification limit. The specificity study was conducted by evaluating the method's capacity to detect and analyze various other components (degradant, excipients, impurities, etc.)
Result and Discussion
Preliminary Analysis
The preliminary experiments were carried out per previously published literature to develop a chromatography method for estimating ERL stability. A variety of mobile phase combinations were tested using acetonitrile and methanol as an organic phase with different proportions of water at a flow rate ranging from 0.7 to 1.1 mL/min. The findings emphasize that degradation peaks separation from the drug-required buffer. The ERL pKa value was used to determine the buffer pH, as effective buffer pH should be within two units of drug pKa [38]. After collecting all the preliminary assessment data, CQA, CPP and CMAs were listed and further studied in detail.
Plackett–Burman Screening Design
The listed CQA, CPP, CMAs (Table S1) were used to prepare the Ishikawa (Fishbone) diagram (Fig. 2). Further ICH Q9 concept is implemented for risk and risk assessment, as shown in Fig S1. A systemic risk assessment identifies essential techniques and crucial process parameters whose variability may affect possible CQAs. Screening designs are suitable when multiple independent factors are expected to influence a response variable. The goal of this study was to employ a Placket Burman screening design to identify the important parameters influencing CAAs. Each selected variable posed a high, medium, or low relative risk (quality characteristic has a large impact, medium or low impact). The primary effects of the eleven selected independent variables were studied on selected CAAs from the Ishikawa diagram (Fig. 2) using the Plackett Burman design. The design has a two-level multifactorial design that can be used to identify the significant independent variables using some short trials [39]. It is used to identify independent factors that need to optimize and critically taken care of during the experiment. The selected eleven factors (Mobile phase composition (ACN: Buffer; v/v), pH of buffer, flow rate, column temperature, degradation sample, type of buffer, wavelength, length of column, type of organic phase, column type, and stressor) studied at two levels [low (− 1) and high (+ 1)] from preliminary trials as shown in Table 1. The preliminary observations show that methanol with ERL caused a variation instability, indicating that the type of organic phase in the mobile phase composition as a categorical component, hence investigated at two levels: acetonitrile (ACN) and methanol (MeOH). The green stability method can develop by using a higher proportion of aqueous components in the mobile phase. Hence, the different proportion of ACN: Water tried but the observed need of buffer for appropriate separation and Gaussian peak. Further, the buffer selection is also crucial for the LC–MS/MS-compatible method, and having that aim studied selected two volatile buffers, ammonium acetate (AA) and ammonium formate (AF). The ammonium acetate has a pKa of 4.76, and the ammonium formate pKa is 3.74; the buffer pH range was selected in the range of ± 1 pH, based on preliminary results. The mobile phase composition studied at two levels [(35:65, v/v), (− 1); (50:50, v/v), (+ 1)]. The pH of the mobile phase has an impact on the peak separation and elution pattern; hence the levels were chosen as 3.5 (− 1) and 4.0 (+ 1). Mobile phase flow rate studied at 0.7 mL/min (− 1) and 1.1 mL/min (+ 1). The column chemistry based on several cross-linked carbons on silica does alter its polarity, which affects chromatography separation. The van Demeter equation connects the variance per unit length of a separation column to the linear mobile phase velocity by analyzing a separation column’s physical, kinetic, and thermodynamic aspects. Hence studied column type [C18 (− 1); C8 (+ 1)], column size [150 cm (− 1); 250 cm (+ 1)] having fixed diameter. The column diameter is an obvious approach to reducing the amount of organic solvent to develop the green method but based on the availability of comparatively polar (C8; Kromasil 100–5-C8, 4.6 × 250 mm, 5µ) and nonpolar (C18; Shimadzu GIST, 4.6 × 250 mm, 5µ) columns selected. C18 has a bigger surface area for the mobile phase to traverse due to the more carbons. This gives the bound phase, and the elutes more time to interact.

Ishikawa Fish-bone diagram for ERL stability indicating method
As a result, the sample elutes more slowly and separates more. At the same time, the C8 column (also known as octadecyl) has a shorter retention duration and sharper peaks. The peak shape can be modified with different peak modifiers hence acetic acid (AA), and formic acid (FA) were selected for the study. The absorbance and peak purity of the drug and its degradation product depends on detection wavelength; hence chosen based on ERL wavelength maxima and tested at 220 nm and 250 nm. The column temperature affects mobile phase viscosity leads to a change in retention time through the change in adsorption energy, as proved by Van’t Hoff by the equation Lnk = ((− ΔH°)/RT) + ((ΔS°)/R) + ln φ where k is retention factor, S is the entropy of reaction between compound and stationary phase, H is the enthalpy of reaction between compound and stationary phase, T is thermodynamic temperature, R is the universal gas constant, and φ is constant relating to phase ratio [40]. Also, using a higher column temperature will result in the aqueous component of the mobile phase behaving more like an organic solvent as the dielectric constant increases, consequently decreasing the amount of organic solvent required. Hence, we studied at 10 °C (− 1) and 40 °C (+ 1). All above factors at two-level tested with 6 Hrs and 12 Hrs degradation sample as shown in Table 1.
The collected sample was studied with HPLC and analyzed statistically with the Design expert v13. The significant variables were selected using the Pareto chart, as shown in Fig. S2. It shows that the length of each bar in the Pareto chart is equivalent to the magnitude of the regression coefficient of that element, which are derivatives of multivariate regression analysis. The statistical parameters, Bonferroni limit, and t-value were used to select significant secondary parameters which need to optimize further. The statistical parameters reveal that the effect of degradation sample, detection wavelength, stressor concentration, column type, column length, and column temperature is less significant. Therefore, these factors had to keep constant for further study. According to the conventional Pareto ranking analysis, mobile phase composition, flow rate, buffer pH, and buffer type significantly impacted method CAAs. As a result, these variables were chosen as CMPs, which need to study in-depth for analytical optimization using design expert v13 response surface design.
The main factor effect, factors interaction and selected variables impact on response variables studied with the desirability function.
Method Optimization by Central Composite Design
As an outcome of secondary parameter screening by Plackett Burman, mobile phase composition, pH of the buffer, flow rate, and buffer type are critical factors that need to be optimized further. The optimization design was selected using the expert v13 response surface matrix with consideration of fewer experimental runs without loss of the orthogonal nature of the model. The selected four critical parameters were optimized using the central composite design (CCD) with n = 30 experiments. The response surface design is employed to optimize four components for the three levels. Selected factors studied in the range from low to high levels as shown in Table 2. The selected four critical factors were mobile phase composition (A), buffer pH (B), flow rate (C), and buffer type (D). The selected-response variable was the resolution of degradation peaks from drug peaks. The goal was to study the factor's primary effect, interaction effect, and quadratic impacts of critical factors on selected response variables RS 1, RS 2, RS 3, RS 4, and RS 5, where RS 1 means resolution between degradation peak 1 and 2. The listed 30 trials, as shown in Table 3, were performed and collected results of resolution studied for numerical and graphical optimization. The mathematical model was developed for each response variable (RS 1, RS 2, RS 3, RS 4, and RS 5), and statistical calculations using ANOVA results were performed. The statistical values for model p-value, R2, and adjusted R2 were studied to clarify the significance of the model, as shown in Table S3. The calculated R2 for all response variables ranged around 0.9, indicating that the models could account for a significant variation in the responses. The summary statistics from the mathematical model (quadratic) demonstrated a reasonably good agreement between the adjusted R2 and R2 values for all response variables. The response variables are expressed in factor addition and subtraction in the polynomial equation. The coded polynomial equation and the p-value imply that four factors substantially affect each response variable. The fitted model was relatively suitable for the experimental results, as shown in Table S3, with a p-value less than 0.05 and a lack-of-fit greater than 0.05.
The design generates the polynomial quadratic equation, as shown in Table S3.
From the above equation, it was observed that factors A, C, BC, BD, CD, A2, and B2 have a negative effect, while B, D, AB, AC, AD, and C2 have a positive impact on the resolution between peaks. Negative value coefficients of A, C, BC, BD, CD, A2, and B2 factors indicate that separation of degradation peaks is inversely proportional to change in factor proportion. The positive sign of coefficients of B, D, AB, AC, AD, and C2 factor indicates that the separation of degradation peaks is directly proportion to the change in factor proportion.
The statistically significant model was used for numerical and graphical optimization. The numerical optimization using software gives several predictions having different desirability. The sample was injected into conditions suggesting desirability 1 to compare the difference between predicted (software) and actual resolution (shown in Fig. 3). As a result, parameters like mobile phase composition (X1) and pH (X2) were selected as critical method parameters for optimization. At the same time, less significant factors flow rate (X3) and buffer type (X4) were set to 0.9 mL min−1 and ammonium formate (AF), respectively. The counterplot derived for each response variable, as shown in Fig S3, represent the effect of factor X1 and X2 on the resolution as the response variable.

Desirability plot for the ERL stability indicating method
Further graphical optimization is done by implementing constraints. The design space (Fig. 4) shows the robust working area, which fulfills the stability-indicating method's criteria of having a resolution of more than 1.5 between peaks. The yellow zone in an overlay plot indicates ideal method circumstances provided by design expert software to achieve method targets. This is the most reliable and operable region for the ERL stability-indicating method. The optimized condition was mobile phase composition of [(42:58; v/v) (ACN: 15 mM Ammonium formate pH 3.8 adjusted with formic acid, the flow rate of 0.9 mL/min. The desirability of 1.0 was the best condition which needs to confirm with practical (Fig. 5). The estimated optimum solution was located within operable analytical design space, and graphical optimization agreed with numerical optimization. The experimental work results employed to reassess risk analysis of factors and updated risk assessment data show all have moderate to low risk. The updated risk assessment (Fig. 6) demonstrates the use of a systemic approach, with high severity factors being converted into lower severity factors.

Design space for the ERL stability indicating method

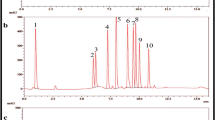
Overlay Chromatogram for i acid, alkaline and oxidative hydrolysis, ii thermal, photolytic and neutral hydrolysis

Updated risk assessment for ERL stability method
Control Strategy
The control strategy is considered the last step in the implementation of QbD. It explains the defined acceptable ranges for each analyzed factor, which impacts drug stability and critical process parameters. All the factors proposed range decided based on results and statistical calculation. The control strategy does help scientists monitor the robustness of the method, as shown in Table 4.
The optimized chromatogram and chromatographic conditions were used to analyze the drug’s stability and degradation in all degradation conditions. The ERL was studied in all degradation conditions (acid hydrolysis, alkaline hydrolysis, peroxide degradation, photolytic degradation, and thermal degradation) and overlay taken as shown in Fig. 3. The six DPs in different degradation conditions and observed drug degradation is shown in Table 5. The Rt (retention time) of formed degradation products in various conditions was used to assign a nomenclature. The optimized condition resolves the ERL from six degradation products with proper and adequate resolution in different degradation conditions, essential for the chemical stability method. The drug was sensitive toward hydrolysis while stable in oxidative, thermal, and photolytic degradation. The found DPs in acid hydrolysis are DP I, II, III, IV, VI, and in alkaline hydrolysis, DP I, II, III, IV, V, and VI.
ERL Degradation Behavior
The ERL studied all degradation conditions with the optimized method and found it is specific, selective, robust, accurate, and precise to separate all degradation product peaks from the drug peak. It was found that ERL is sensitive to acidic and alkaline hydrolysis due to stress degradation. It was confirmed by the additional peaks obtained in the respective chromatography and the decrease in the area of ERL in each condition compared to their area at zero time. The mass balance was calculated using the ERL responses and all of the degradation products obtained from the stress studies at the selected detection wavelength. For all degradation conditions, the mass balance was greater than 80 except for thermal degradation. In thermal degradation, the mass balance was 75% due to loss of mass during degradation. Mass can be lost and reduce the percent degradation because of the release of water in thermal degradation or due to the formation of volatile products or products having no chromophore or diffusive losses into or through containers [4]. The drug stability study reveals that ERL is sensitive toward hydrolysis, which leads to strict monitoring for drug liquid dosage form stability.
Analytical Method Validation
The developed SIAM was validated according to ICH recommendations Q2 (R1), as shown in Tables 6 and 7. The results show that the % RSD values for all parameters were less than one. The regression coefficient of 10–50 μg/mL was 0.9983, which confirms the method’s linearity. The optimized method could separate all possible degradation peaks from the drug peak with resolution (> 2) and peak purity ranging from 0.990 to 0.999. LOD and LOQ values were 0.366 μg/mL and 1.1 μg/mL, respectively. The precision (Interday and intraday) was performed at three concentrations, including 10, 30, and 50 µg/mL (each in triplicate) on three consecutive days and on the same day; observed RSD values were 0.225 and 0.144, respectively. The accuracy % Recovery RSD was estimated to be (100.45 ± 0.2110–102.77 ± 0.4396), and the % Assay RSD was found (100.45–102.77) ± 0.998.
Method Greenness Score
The procedure involves external sample treatment with a reduced number of steps on HPLC. The sample injected into HPLC has a run time of 40 min with a flow rate of 0.9 mL/min. The developed method is an isocratic having solvent composition as ACN (42% v/v): ammonium formate 15 mM (58% v/v). The green rating is an interesting approach to scoring the green credentials of an HPLC method. The output from AGREE tool is a clock-like graph, with the overall score and color representation used to calculate method greenness (Fig. 7). The overall score is 0.72, with a value close to 1 and a dark green color indicating that the assessed method is greener than other published methods. The color in the segment with the number corresponding to each criterion reflects the procedure's performance in each assessment criteria [17, 41].

ERL stability indicating method greenness assessment
The input criteria are based on the 12 SIGNIFICANCE principles, and different weights are assigned to them, allowing for some flexibility. Sample treatment, sample amount, device location, sample preparation stages, automation/miniaturization, derivatization, waste, analysis throughput, energy consumption, source of reagents, toxicity, and operator safety are the 12 SIGNIFICANCE [42]. The Principles of Green Analytical Chemistry translate each of the 12 input variables into a common scale in the 0–1 range. The sum of the assessment results for each principle is the final assessment result. The result is a clock-like graph in the middle, with the overall score and color representation (see Fig. 7). The intuitive red-yellow-green color scale reflects the procedure's performance in each principle, while the width of its associated section expresses the weight of each principle. The assessment can be completed quickly and easily with the help of user-friendly software, which includes an automatically generated graph and a report [18, 19, 41]. The overall score is depicted in the middle of the pictogram, with numbers near 1 and a dark green color indicating that the evaluation technique is greener. The color in the segment with the number corresponding to each criterion reflects the procedure’s performance in each assessment criteria. As shown in Fig. 7, the scores corresponding to GAC principles 1, 7, and 10 are quite low, while, in the case of principles 2, 3, 4, 5, 6, 11 and 12, the performance is excellent [36].
Conclusion
Regulatory agencies and pharmaceutical firms are increasingly focusing on quality terms, making continual monitoring of each parameter throughout each stage even more essential for companies. The present study effectively shows the effectiveness of the quality by design approach in optimizing the ERL stability-indicating chromatographic technique and deeper understanding of the significant factor related to method performance. The Analytical Quality by Design (AQbD)-driven method was found sensitive, selective, accurate, and robust to separate six DPs in different degradation conditions. This unique method assists the analyst in defining control strategies to reduce the negative impact of these CMVs on method performance. The validation studies revealed that the system has outstanding linearity, accuracy, precision, sensitivity, robustness, and ruggedness. The drug (ERL) was sensitive under hydrolytic (acid and alkali) conditions while stable under oxidative, thermal, and photolytic conditions; henceforth, strict stability monitoring during liquid dosage formulation is suggested. AQbD driven method development leads to having LC–MS-compatible green method. The developed stability-indicating approach may be used to track and monitor ERL stability in its different formulations and for bioanalytical research as it is mass compatible. The initiative also seeks to increase environmental awareness to develop solutions with a practical, green, and comparatively cost-effective development method. The AGREE tool was used to illustrate the method of greenness. The developed method found green, LC–MS compatible, and sensitive.
Availability of Data
All the data generated or analyzed during this study are included in this published article and in form of supplementary data.
Abbreviations
- nm:
-
Nanometer
- ppm:
-
Parts per million
- °C:
-
Degree Celsius
- mL:
-
Milliliter
- µg:
-
Micron gram
- SIAM:
-
Stability indicating assay method
- mg:
-
Milligram
- Conc.:
-
Concentration
- HPLC:
-
High performance liquid chromatography
- LC/MS–MS:
-
Liquid Chromatography Mass Spectrometry
- HCl:
-
Hydrochloric acid
- NaOH:
-
Sodium hydroxide
- MeOH:
-
Methanol
- ACN:
-
Acetonitrile
- pH:
-
Potential of hydrogen
- Hrs:
-
Hours
- min:
-
Minute
- DOE:
-
Design of experiment
- QbD:
-
Quality by design
- ERL:
-
Erlotinib
- H2O2 :
-
Hydrogen peroxide
- PBD:
-
Plackett Burman design
- CCD:
-
Central composite design
- RSD:
-
Response surface design
- ANOVA:
-
Analysis of variance
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- ICH:
-
International Council for Harmonization
- DPs:
-
Degradation products
- CQAs:
-
Critical quality attributes
- CAAs:
-
Critical analytical attributes
- AMGS:
-
Analytical method greenness score
- CMAs:
-
Critical material attributes
References
Blessy M, Patel RD, Prajapati PN, Agrawal YK (2014) Development of forced degradation and stability indicating studies of drugs—a review. J Pharm Anal 4:159–165. https://doi.org/10.1016/J.Jpha.2013.09.003
Bajaj S, Singla D, Sakhuja N (2012) Stability testing of pharmaceutical products. J Appl Pharm Sci 2:129–138. https://doi.org/10.7324/JAPS.2012.2322
Baber N (1994) International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH). Br J Clin Pharmacol 37(5):401. https://doi.org/10.1002/9781118445112.stat07000
Bakshi M, Singh S (2002) Development of validated stability-indicating assay methods—critical review. J Pharm Biomed Anal 28:1011–1040. https://doi.org/10.1016/S0731-7085(02)00047-X
Hirsh V, Thongprasert S, Campos D et al (2005) Erlotinib in previously treated non-small cell lung cancer. New Eng J. https://doi.org/10.1056/NEJMoa050753
Bareschino MA, Schettino C, Troiani T, Al Et (2007) Erlotinib in cancer treatment. Ann Oncol 18:35–41. https://doi.org/10.1093/Annonc/Mdm222
Field JK, Smith RA, Aberle DR, Al Et (2012) International association for the study of lung cancer computed tomography screening Workshop 2011 Report. J Thorac Oncol 7:10–19. https://doi.org/10.1097/JTO.0b013e31823c58ab
Fry WA, Menck HR, Winchester DP (1996) The national cancer data base report on lung cancer. Cancer 77:1947–1955. https://doi.org/10.1002/(SICI)1097-0142(19960501)77
Tóth G, Jánoska Á, Szabó ZI, Al Et (2016) Physicochemical characterisation and cyclodextrin complexation of erlotinib. Supramol Chem 28:656–664. https://doi.org/10.1080/10610278.2015.1117083
Diven DG, Bartenstein DW, Carroll DR (2015) Extending shelf life just makes sense. Mayo Clin Proc 90:1471–1474. https://doi.org/10.1016/J.Mayocp.2015.08.007
International Committee Harmonization (2018) International council for harmonisation of technical requirements for pharmaceuticals for human use final concept paper ICH Q14: analytical procedure development and revision of Q2(R1) analytical validation. https://database.ich.org/sites/default/files/ICH_Q14_Document_Step2_Guideline_2022_0324.pdf
Psimadas D, Georgoulias P, Valotassiou V, Loudos G (2012) Molecular nanomedicine towards cancer. J Pharm Sci 101:2271–2280. https://doi.org/10.1002/Jps.23146
Monks K, Molnár I, Rieger HJ, Al Et (2012) Quality by design: multidimensional exploration of the design space in high performance liquid chromatography method development for better robustness before validation. J Chromatogr A 1232:218–230. https://doi.org/10.1016/J.Chroma.2011.12.041
Kumar PA, Kumar YR, Jayashree A (2016) Development and validation of a stabilityindicating RP-HPLC method for the estimation of erlotinib impurities by Qbd approach. Rasayan J Chem 9:180–188. https://doi.org/10.1009/76-0083rjc.2016
Orlandini S, Pinzauti S, Furlanetto S (2013) Application Of quality by design to the development of analytical separation methods. Anal Bioanal Chem 405:443–450. https://doi.org/10.1007/S00216-012-6302-2
Kim MK, Park SC, Park G et al (2021) Analytical quality by design methodology for botanical raw material analysis: a case study of flavonoids In Genkwa Flos. Sci Reports 111(11):1–14. https://doi.org/10.1038/S41598-021-91341-W
Hicks MB, Farrell W, Aurigemma C et al (2019) Making the move towards modernized greener separations: introduction of the analytical method Greenness Score (AMGS) calculator. Green Chem 21:1816–1826. https://doi.org/10.1039/C8gc03875a
Pena-Pereira F, Wojnowski W, Tobiszewski M (2020) AGREE—analytical greenness metric approach and software. Anal Chem 92:10076–10082. https://doi.org/10.1021/Acs.Analchem.0c01887
Saroj S, Shah P, Jairaj V, Rathod R (2018) Green analytical chemistry and quality by design: a combined approach towards robust and sustainable modern analysis. Curr Anal Chem 14:367–381. https://doi.org/10.2174/1573411013666170615140836
Gamal M, Naguib IA, Panda DS, Abdallah FF (2021) Comparative study of four greenness assessment tools for selection of greenest analytical method for assay of hyoscine: N-Butyl Bromide. Anal Methods 13:369–380. https://doi.org/10.1039/D0ay02169e
Naz A, Tabish I, Naseer A et al (2021) Green chemistry approach: method development and validation for identification and quantification of entecavir using FT-IR in bulk and pharmaceutical dosage form. Futur J Pharm Sci 71(7):1–8. https://doi.org/10.1186/S43094-021-00211-9
Latha ST, AnandaThangadurai S, Jambulingam M et al (2017) Development and validation Of RP-HPLC method for the estimation of erlotinib in pharmaceutical formulation. Arab J Chem 10:S1138–S1144. https://doi.org/10.1016/J.Arabjc.2013.02.006
Mahajan AA, Miniyar PB, Patil AS et al (2015) Separation, identification, and characterization of degradation products of erlotinib hydrochloride under ICH-recommended stress conditions by LC, LC-MS/TOF. J Liq Chromatogr Relat Technol 38:629–639. https://doi.org/10.1080/10826076.2014.936610
Babu C, Narasimha Rao KL, Devanna N, Suresh Reddy KVN (2016) Development and validation of a stability indicating HPLC method for the quantification of impurities in erlotinib hydrochloride dosage forms. Int J Res Pharm Sci 7:98–105. https://doi.org/10.10863/sc/583.2016
Pujeri SS, Khader AMA, Seetharamappa J (2009) Validated stability-indicating chromatographic method for the assay of erlotinib active pharmaceutical ingredient. Anal Lett 42:1855–1867. https://doi.org/10.1080/00032710903061170
Government of India, Ministry of Health (2018) The Indian Pharmacopeia Volume-II, Indian Pharmacopeia Commission Ghaziabad, 1969
Sharma G, Thakur K, Raza K, Katare OP (2019) Stability kinetics of fusidic acid: development and validation of stability indicating analytical method by employing analytical quality by design approach in medicinal product(S). J Chromatogr B 1120:113–124. https://doi.org/10.1016/J.JCHROMB.2019.05.001
Tome T, Žigart N, Časar Z, Obreza A (2019) Development and optimization of liquid chromatography analytical methods by using aqbd principles: overview and recent advances. Org Process Res Dev 23:1784–1802. https://doi.org/10.1021/ACS.OPRD.9B00238
Sahu PK, Ramisetti NR, Cecchi T, Al Et (2018) An overview of experimental designs In HPLC method development and validation. J Pharm Biomed Anal 147:590–611. https://doi.org/10.1016/J.JPBA.2017.05.006
Yao H, Vancoillie J, D’Hondt M, Al Et (2016) An analytical quality by design (Aqbd) approach for A L-asparaginase activity method. J Pharm Biomed Anal 117:232–239. https://doi.org/10.1016/J.JPBA.2015.08.042
Teng J, Zhu C, Lyu J et al (2022) Analytical lifecycle management (ALM) and analytical quality by design (Aqbd) for analytical procedure development of related substances in tenofovir alafenamide fumarate tablets. J Pharm Biomed Anal. https://doi.org/10.1016/J.JPBA.2021.114417
International Committee Harmonization (2015) International council for harmonization of technical requirements for pharmaceuticals for human use, ICH guideline Q9 on quality risk management. https://www.ich.org/page/quality-guidelines
Suryawanshi D, Jha DK, Shinde U, Amin PD (2019) Development and validation of a stability-indicating RP-HPLC method of cholecalciferol in bulk and pharmaceutical formulations: analytical quality by design approach. J Appl Pharm Sci 9:21–32. https://doi.org/10.7324/JAPS.2019.90604
Shinde DD, Ahirrao S, Prsad R (2018) Fishbone diagram: application to identify the root causes of student–staff problems in technical education. Wirel Pers Commun 1002(100):653–664. https://doi.org/10.1007/S11277-018-5344-Y
Patel MN, Kothari CS (2021) A comprehensive stability study of vardenafil using quality by design approach. Chromatogr 848(84):751–767. https://doi.org/10.1007/S10337-021-04059-2
Kurmi M, Kumar S, Singh B, Singh S (2014) Implementation of design of experiments for optimization of forced degradation conditions and development of a stability-indicating method for furosemide. J Pharm Biomed Anal 96:135–143. https://doi.org/10.1016/J.Jpba.2014.03.035
Youn BD, Choi KK (2004) A new response surface methodology for reliability-based design optimization. Comput Struct 82:241–256. https://doi.org/10.1016/J.Compstruc.2003.09.002
Snyder LR, Kirkland JJ, Glajch JL (2012) Basics of separation. Pract HPLC Method Dev. https://doi.org/10.1002/9781118592014.CH2
Vanaja K, Rani RHS (2007) Design of experiments: concept and applications of Plackett Burman design. Clin Res Regul Aff 24:1–23. https://doi.org/10.1080/10601330701220520
Lima EC, Gomes AA, Tran HN (2020) Comparison of the nonlinear and linear forms of the Van’t Hoff equation for calculation of adsorption thermodynamic parameters (∆S° And ∆H°). J Mol Liq 311:113315. https://doi.org/10.1016/J.MOLLIQ.2020.113315
Gamal M, Naguib IA, Panda DS, Abdallah FF (2021) Comparative study of four greenness assessment tools for selection of greenest analytical method for assay of hyoscine N-butyl bromide. Anal Methods 13:369–380. https://doi.org/10.1039/D0AY02169E
Kelani KM, Elzanfaly ES, Saad AS, Al Et (2021) Different greenness assessment perspectives for stability-indicating RP-HPLC method used for the assay of isoxsuprine hydrochloride and four nephrotoxic and hepatotoxic photothermal degradation products. Microchem J 171:106826. https://doi.org/10.1016/J.MICROC.2021.106826
Funding
The authors have not received any fund for the work.
Author information
Authors and Affiliations
Contributions
All authors compiled, read and approved the final version of the manuscript for publication.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gundecha, S., Patel, M. & Mayur, Y.C. An Application of Quality by Design and Analytical Greenness Assessment Approach for the Development of Erlotinib Stability Indicating Method. Chromatographia 85, 575–588 (2022). https://doi.org/10.1007/s10337-022-04167-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-022-04167-7