
Abstract
An analytical method was developed for the determination of 16 polycyclic aromatic hydrocarbons (PAHs) and 15 nitrated PAHs (NPAHs) in ambient air particulates by applying an in-house built integrated ultrasonic extraction device followed by gas chromatography coupled with mass spectrometry (GC–MS). After integrated ultrasonic extraction in the extraction cell, the extract solution was released through the opened valve into a purification tube containing adsorbents for purification, then concentrated and analyzed by GC–MS. Compared to previously reported methods, such as Soxhlet extraction and accelerated solvent extraction, this method has greatly shortened the pretreatment time, simplified the pretreatment process, and reduced experimental costs. A series of experimental conditions, including extraction solvent, adsorbents, and elution solvent, were systematically studied and optimized. Owing to its superior ability in purifying particulate matter samples, a combination of flower-like-texture Mg–Al layered double oxide (Mg–Al-LDO) and silica gel was selected as the optimized adsorbent. To our knowledge, few reports on this combination of materials exist. The limits of detection (LODs) of 16 PAHs and 15 NPAHs ranged from 0.018 to 1.7 and from 0.035 to 0.60 ng g−1, respectively. Limits of quantifications (LOQs) were from 0.061 to 5.7 and 0.12 to 2.0 ng g−1 for 16 PAHs and 15 NPAHs, respectively. Recoveries at three spiked concentration levels ranged from 81.6 to 116%. The relative standard deviations (RSDs) were less than 6.5% in all cases. The proposed analytical method was proved to be practical and suitable technique for PAH and NPAH analysis in real ambient air particulate samples.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a category of organic pollutants ubiquitous in the atmospheric environment. They are generated by incomplete combustion processes, such as domestic coal burning and industrial cooking [1, 2]. After emission into the atmosphere, a series of reactions, e.g., oxidation and nitration can produce derivatives of PAHs, such as nitrated polycyclic aromatic hydrocarbons (NPAHs), which alters the properties of PAHs, including their toxicity. The US Environmental Protection Agency (US EPA) has already listed 16 PAHs as priority pollutants; moreover, European ambient air legislation has recommended 1 ng m−3 as a guideline value for the annual average concentration of benzo(a)pyrene not to be exceeded [3, 4]. NPAHs are emitted into the environment with PAHs, which contain at least one nitro-functional group on the aromatic benzene ring of a PAH. Because most NPAHs show higher mutagenicity and carcinogenicity than their precursors [5], which are also used as indicators in tracing air contamination, NPAHs have been monitored and investigated in research work in recent years, although there are no regulations for NPAHs to date [4].
In ambient air particulates, PAHs and NPAHs are usually analyzed by solvent extraction, and then high-performance liquid chromatography with fluorescence detection (HPLC–FLD) [3, 6], high-performance liquid chromatography with tandem mass spectrometry (HPLC–MS/MS) [8] or gas chromatography with mass spectrometry (GC–MS) [e.g., electron impact ionization (EI) mode for PAHs or negative chemical ionization (NCI) mode for NPAHs] [7, 9,10,11,12] analysis. Soxhlet extraction [13, 14], accelerated solvent extraction (ASE) [9, 15,16,17], supercritical fluid extraction [18], ultrasonication [9, 19], and microwave extraction [3, 20] have mostly been used for the extraction of nitrated polycyclic aromatic hydrocarbons (NPAHs) or PAHs. Prior to instrumental analysis, a cleanup procedure is necessary for sample extraction to reduce matrix effects during analysis. Silica cartridges [5, 12], alumina cartridges [13], combined silica-alumina cartridges [9, 20], aminopropyl cartridges [10], HPLC (normal phase) [21], carbon nanotubes [18], and QuEChERS-like extraction [11] have been chosen as cleanup techniques for ambient air particulates extraction according to the literature. Although all of these sample preparation methods are applicable to the determination of organic contamination in ambient air particulates, they are usually time-consuming, costly, labor intensive, and require large volumes of organic solvent [22,23,24]. Considering the actual needs of atmospheric environmental monitoring work, a simple, effective, and low-cost sample preparation technique is desired for the determination of PAHs and NPAHs in ambient air particulates.
An integrated ultrasonic simultaneous extraction and cleanup system is presented in this paper. This system combines integrated ultrasonic extraction and purification in one step, which avoids the sample-transfer steps and saves solvent and time. A novel flower-like Mg–Al layered double oxide (Mg–Al-LDO) with silica gel, instead of the traditional sorbent [9, 20], was used in the purification tube for the pretreatment procedure. Mg–Al-LDO is a composite oxide compound of magnesium and aluminum [25] that has much more polar surface area owing to the presence of magnesium, which is known to adsorb polar impurities from tea and sediment for determining pesticide and dechlorane content [26, 27]. In this research, the flower-like Mg–Al-LDO material, which possesses a large surface area due to its special morphology, was synthesized and implemented as an adsorbent for the analysis of PAHs and NPAHs in an ambient air particulate matrix. The detailed description of this system is discussed herein. Each sample required only about 30 min of pretreatment time using this proposed method. However, for ASE extraction, heating up time of extraction furnace was about 6 min. The ASE extraction procedure, including static time, flushing, and purging, required at least 20 min. After ASE extraction, sample extract was transferred to flask for evaporating and blown by nitrogen before SPE purification, which also needed at least 20 min. Then SPE purification procedure needed at least 10 min. From start to finish, the ASE extraction and sample pretreatment time were usually about 56 min. So, this proposed method has greatly shortened the pretreatment time, simplified the pretreatment process, and reduced experimental costs.
Experimental
Chemicals, Reagents, and Instruments
The chemicals, solvents, and instruments used in the experiments are provided in the Electronic Supplementary Material.
Synthesis and Characterization of Flower-Like Mg–Al-LDH and Flower-Like Mg–Al-LDO
The adsorbent of flower-like Mg–Al-LDO was synthesized using a microemulsion system. This approach provided an excellent method of controlling the morphology of layered double hydroxide (LDH) [28]. The details of the synthesis and characterization are provided in the Electronic Supplementary Material.
Sampling
The total suspended particulate (TSP) samples, which were collected from the top of a six-story building in Beijing, China (39°58″30′N, 116°25″49′E) in June and December 2015. All samples were collected on glass fiber filters (GFFs; 47 mm i.d., Pall Co., New York, USA) over 44 h at a flow rate of 100 L min−1 using a medium-volume sampler (KC-6120, Qingdao Laoshan Electronic Instrument Co., Qingdao, China). Detailed information on the TSP samples is listed in Electronic Supplementary Material Table S1. The GFFs were heated at 500 °C in a muffle furnace (FP31, Yamato, Japan) for 4 h to remove all organic pollutants, equilibrated at 25 °C in a desiccator for 24 h, and then weighed. After collection, filters were wrapped in annealed aluminum foil, sealed, and stored at −18 °C before analysis.
Purification Tube Preparation
In this study, the purification tube (6 mL capacity, Supelco, Bellefonte, PA, USA) was filled with two adsorbent layers. The upper layer was 500 mg of silica gel, and the lower layer was 500 mg of flower-like Mg–Al-LDO. A piece of glass filter paper was used to plug the tube and retain the sorbents.
Extraction and Purification
ASE Extraction Method
For ASE (ASE350, Dionex, California, USA) extraction, 11 mL cells were used under the following extraction conditions: 120 °C, 140 bars (14 MPa), two cycles of 5 min duration each for heat and static times, 90% flushing, and purging for 100 s. The first extraction was performed with dichloromethane and the second with acetone/DCM (2:1 v/v). The above conditions were previously used to extract PAHs, NPAHs, or oxygenated PAHs [29,30,31].
Extraction and Purification Using the Proposed Method
The integrated ultrasonic extraction system consists of three parts: an ultrasonic grinder instrument (VCX 130, Sonics Co., CT USA), an extraction cell, and a purification tube (as shown in Fig. 1). The 35 mL extraction cell (Comma Biological Technology Co., Guangzhou, China) was linked to the purification tube by a flow-control valve and a tube adapter. A quarter of TSP filter sample was cut and placed in the extraction cell. To the extraction cell, 15 mL of hexane/DCM (1:1, v/v) solvent mixture were added for ultrasonic extraction. An alloy probe was put into the extraction cell for 20 min of extraction. After ultrasonication, the extract was directly passed through the opened control valve into the purification tube for cleanup. From the upper extraction cell, 12 mL of hexane/DCM (3:7, v/v) were added to wash the system and elute the analytes (Fig. 1). The eluate was collected and concentrated to 0.2 mL by a moderate nitrogen flow before GC–MS analysis.

In-house built integrated ultrasonic extraction device
GC–MS Analysis
GC–EI–MS Analysis
The GC–MS instrument (Agilent 6890N-5975C, Agent Technologies, Santa Clara, CA, USA) with an electron impact ionization (EI) ion source was used to analyze 16 PAHs. The GC injection port was kept at 280 °C and a sample (1 µL) was injected in splitless mode with splitless time of 1 min. Target compounds were separated by a 50-m DB-5MS column (0.25-mm ID, 0.1-µm film thickness; J&W Scientific, Folsom, CA, USA). The carrier gas was helium (99.999%) at a constant flow rate of 1 mL min−1. The GC oven temperature program was as follows: held at 60 °C for 0.5 min; 30 °C min−1 to 200 °C; 3 °C min−1 to 225 °C; 6 °C min−1 to 266 °C; 4 °C min−1 to 300 °C, and held for 30 min. The transfer-line and ion-source temperatures were set at 280 and 230 °C. The MS was run in selected ion monitoring (SIM) mode. Monitored quantitative and qualitative ions for all analytes are listed in Table 1.
GC–NCI–MS Analysis
NPAHs were analyzed by a GC–MS instrument (Agilent 7890A-5975C, Agilent Technologies, Santa Clara, CA, USA) with negative chemical ionization (NCI), which possesses higher selectivity (in addition to sensitivity) for NPAHs. The GC injection port was kept at 280 °C. A sample (1 µL) was injected in splitless mode with a pressure pulse of 25 psi for 1.5 min. Target compounds were separated by a 15-m DB-5HT column (0.25-mm I.D., 0.1-µm film thickness; J&W Scientific, Folsom, CA, USA). The carrier gas was helium (99.999%) at a constant flow rate of 1 mL min−1. The flow rate of methane (99.999%) was 2.5 mL min−1. The GC oven temperature program was as follows: held at 60 °C for 0.5 min; 8 °C min−1 to 220 °C; 20 °C min−1 to 300 °C, and held for 15 min. The transfer-line and ion-source temperatures were set at 300 and 230 °C. The MS was run in SIM mode. Monitored quantitative and qualitative ions for all analytes are listed in Table 1.
Results and Discussion
Optimization of Extraction Conditions
Extraction Solvent
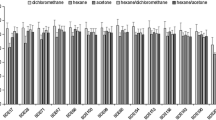
To achieve effective extraction, the collected TSP filter samples, rather than spiked filters, were applied to optimize the extraction solution. The ASE extraction method [29,30,31] was used as a reference. To reduce the uncertainty caused by different purification processes, the same cleanup procedure was used and the extraction time was uniformly 20 min in this section. The extract was purified using the double-layered absorbent mentioned above with 12 mL of hexane/DCM (3:7, v/v). One single solvent (hexane) or solvent mixtures (hexane/DCM (1:1, v/v) or hexane/acetone (1:1, v/v) were evaluated to determine their applicability as extraction solvents for ultrasonic extraction. A sampling filter was divided into four equivalent pieces. One piece of filter was extracted by ASE method and the other three were extracted with the three solvent using an ultrasonic probe. The results are shown in Fig. 2. Overall, the masses of PAHs (Fig. 2a) and NPAHs (Fig. 2b) extracted by hexane/DCM (1:1, v/v) or hexane/acetone (1:1, v/v) were similar. The extraction efficiency of PAHs and NPAHs in hexane was much lower than that of the other two extraction solvents. Compared to the result obtained using the ASE method, there was also no obvious difference in extraction efficiency using ultrasonic extraction with the two mixed solvents. Hexane/DCM (1:1, v/v) was selected as the optimal extraction solvent because an extraction solvent with acetone extracts more pigments observed obviously that causes interference in the GC–MS analysis.

Comparison of the average a PAH or b NPAH quantity extracted according to the extraction solvent used [hexane, hexane/DCM (1:1, v/v), or hexane/acetone (1:1, v/v)] using the ultrasonic procedure and ASE method. Filter sections originated from the same TSP filter samples (n = 3). For both extraction methods, the same cleanup procedure was used
Extraction Time
The influence of extraction time on extraction efficiency of PAHs and NPAHs in the ultrasonic extraction procedure was investigated. The results obtained are shown in Fig. 3a, b. Using 15 mL of hexane/DCM (1:1, v/v) as an extraction solvent, the ultrasonic time (10, 20, and 30 min) was studied using the same cleanup method as in the solvent extraction experiment. It was found that 10 min of ultrasonic extraction was not exhaustive. As can be seen (Fig. 3), no significant improvement was observed after 30 min of extraction compared with 20 min extractions. In addition, additional extraction of the filter was performed. The results of this experiment indicate that the target compounds were below the detection limit. Therefore, 20 min was selected as the ultrasonic extraction time.

Comparison of the average a PAH or b NPAH quantity extracted according to the extraction time used (10, 20, and 30 min) using the ultrasonic procedure and ASE method. Filter sections originated from the same TSP filter samples (n = 3). For both extraction methods, the same cleanup procedure was used
Extraction Volume
In the integrated ultrasonic extraction procedure, excess solvent would cause splashing, leading to loss of extraction solution. On the other hand, if the extraction volume was insufficient, the sample filters could not be completely immersed and effectively extracted. To avoid this problem, 15 mL was chosen as the optimal extraction solvent volume.
Cleanup Optimization
To eliminate matrix interference, an effective purification was needed. In this study, the type of adsorbents, the method of filling adsorbents, and the volume and type of elution solvent was evaluated to achieve less matrix interference for TSP extraction. A novel absorbent, flower-like Mg–Al-LDO, was introduced as one of adsorbents for its large polar surface area in the experiment.
Selection of Adsorbent Type
SPE is widely used for purification of extracts of ambient air particulates. Major fillings such as silica gel [5, 12], neutral alumina [13] were used in the SPE column. In this study, four types of adsorbent or their combinations [silica gel, flower-like Mg–Al-LDO, silica gel with flower-like Mg–Al-LDO (1:1, w/w), and silica gel with N-Al2O3 (neutral alumina) (1:1, w/w); 1 g] were investigated for their purification effects. As shown in Fig. 4a, b, when silica gel with flower-like Mg–Al-LDO (1:1, w/w) was used, satisfactory recoveries ranging from 80.3 to 117% and RSDs from 0.53 to 9.7% were achieved for 16 PAHs and 15 NPAHs by contrasting the results from other three adsorbents. Silica gel with flower-like Mg–Al-LDO seemed to show better ability to remove high polar interference, such as pigments, from a sample extract according to Electronic Supplementary Material Fig. S4. The sample purified by silica gel with flower-like Mg–Al-LDO showed the lightest color compared to the three other absorbents. Finally, the combination of silica gel and flower-like Mg–Al-LDO was selected as the adsorbent in this research.

Comparison of sorbents of silica gel, flower-like Mg–Al-LDO (1 g), silica gel + flower-like Mg–Al-LDO (1:1, w/w, 1 g), and silica gel + N-Al2O3 (1:1, w/w, 1 g) on the recoveries of a PAHs and b NPAHs from spiked total suspended particulate samples (n = 3)
Selection of Adsorbent Filling Methods
Since silica gel and flower-like Mg–Al-LDO were chosen as adsorbents, the purification effect using three filling methods in the purification tube with the two adsorbents was investigated. The cleanup tube was filled with mixed adsorbents and flower-like Mg–Al-LDO in the top layer and bottom layer, respectively. The result obtained from the three types of filling methods showed that the extract could not be well purified using mixed adsorbents because of the obvious interference of target analytes in the GC–EI–MS analysis. As for absorbents filled using two-layer methods, there was no obvious interference and no difference in recovery and RSD. The method of filling with flower-like Mg–Al-LDO in the bottom layer was finally chosen because the speed of elution was more easily controlled.
Optimization of Elution Solution
To obtain effective elution, the mixed ratio and volume of hexane and DCM, which were reported as PAH or NPAH eluents [10, 13, 20], were optimized in this study. Because it was almost impossible to obtain blank matrix samples, extracted solutions from ten samples collected in clean air with much lower level of PAH and NPAH were mixed and used as a hypothetical matrix blank. To optimize the elution solvent ratio, half of the mixed sample extract solution was spiked with 1 mL of mixed standard solution containing a concentration of 0.40 and 2.0 µg g−1 for NPAHs and PAHs. Meanwhile, the other half was used for deducting the mass of the analytes in the hypothetical matrix blank. The spiked sample extract was divided into five equal parts, and then eluted by 10 mL of hexane/DCM (4:1, 3:2, 1:1, 3:7, and 0:1, respectively, v/v) solution after loading into the cleanup tube. The same steps were repeated for the other half of the samples without spiking. The results are plotted in Fig. 5, which shows that hexane/DCM (3:7, v/v) was the best choice among five elution solvents, since the recoveries of all target compounds were ranged from 80.6 to 117% and there were no obvious interference peaks. Most of the analytes were difficult to elute using an elution solvent of hexane/DCM (4:1, v/v). When the elution solvents used were hexane/DCM (3:2, v/v) or hexane/DCM (1:1, v/v), the recoveries for PAHs of indeno(1,2,3-cd)pyrene, benzo(g,h,i)perylene, and dibenzo(a,h)anthracene were 47.1–53.2% and 69.9–76.6%, respectively (Fig. 5). However, selecting DCM as elution solvent, many more pigments were eluted and interference peaks were found in the GC–MS determination. Therefore, the solvent mixture of hexane/DCM (3:7, v/v) was used as the eluent in the purification process.

Effect of different elution solvents on the recoveries of a PAHs and b NPAHs from spiked total suspended particulate samples (n = 3)
The elution volume was obtained according to the spiked sample extraction solution. The spiked sample was eluted by 14 mL of hexane/DCM (3:7, v/v) solution, and every 2 mL of eluent was collected and detected. From Electronic Supplementary Material Fig. S5a, b, it was found that the recoveries of most target compounds were near 100% when the eluent volume was 12 mL. Finally, an elution solvent of 12 mL of hexane/DCM (3:7, v/v) was selected as the minimum volume for analytes in the purification procedure.
Matrix Effects
Reducing matrix interference is important to ensure accurate and reliable experimental results. Matrix effects (MEs) could come from the sample matrix, the sample preparation procedure, quality of chromatographic separation, and ionization type [32]. For the complex ambient air particulate matrix, matrix effects have always existed and have become one of the major elements influencing repeatability and accurate quantification in the determination by GC–MS. Using an isotope labeled internal standard method could effectively reduce matrix effects because physical and chemical properties of analytes with corresponding isotope labeled internal standard would be similar [33]. The isotope labeled internal standard method could not only compensate for the change in the target compound response but also efficiently compensate for the loss of the target compounds during the pretreatment procedure [26]. Thus, the isotope labeled internal standard acenaphthylene-d8, phenanthrene-d10, anthracene-d10, fluoranthene-d10, benzo(a)anthracene-d10, benzo(a)pyrene-d10, dibenzo(a,h)-anthracene-d14, 3-nitrobiphenyl-d9, 2-nitrofluorene-d9, and 1-nitropyrene-d9 were used in this study, and matrix effects of internal standard method were evaluated by the following equation [34, 35]:
where A is the chromatographic peak area ratio of the analytes to the internal standard in the matrix standard and B is the chromatographic peak area ratio of the analytes to the internal standard in the calibration standard. Samples with a low level of analytes were used as a hypothetical matrix blank. Therefore, in calculating A in Eq. (1), the value was obtained by deducting the peak area ratio of the hypothetical matrix blank.
ME <0% represents an ionization inhibition, and ME = 0% indicates no matrix effects, while ME >0% indicates an ionization enhancement. The results are listed in Table 2. The matrix effects of 16 PAHs ranged from −8.9 to 12% and those of 15 NPAHs ranged from −14 to 18%, which indicates that matrix interference could be effectively eliminated and compensated by this method.
Evaluation of the Method
Calibration Curve and Linearity
To obtain a good chromatographic separation, chromatographic conditions were optimized for 16 PAHs and 15 NPAHs. The GC–MS method parameters are listed in Table 1. Chromatograms of spiked samples at a concentration of 5.0 × 102 ng g−1 for 16 PAHs and 2.0 × 102 ng g−1 for 15 NPAHs are presented in Electronic Supplementary Material Figs S6 and S7. It was proved that this method was suitable for determination of 16 PAHs and 15 NPAHs in ambient air particulate samples. According to a series of spiked samples, the linearity and linear range of this method could be obtained. The concentrations of prepared calibration standards were between 5.0 and 1.0 × 103 ng g−1 (5.0, 10, 50, 1.0 × 102, 2.5 × 102, 5.0 × 102, and 1.0 × 103 ng g−1) for most PAHs, 10–1.0 × 103 ng g−1 (10, 50, 1.0 × 102, 2.5 × 102, 5.0 × 102, and 1.0 × 103 ng g−1) for indeno(1,2,3-cd)pyrene, benzo(g,h,i)perylene, and dibenzo(a,h)anthracene; and 1.0–5.0 × 102 ng g−1 (1.0, 5.0, 10, 50, 1.0 × 102, 2.5 × 102 and 5.0 × 102 ng g−1) for 15 NPAHs. The limits of detection (LODs) (signal-to-noise ratio of 3) and the limits of quantifications (LOQs) (signal-to-noise ratio of 10) were calculated using the chromatograms obtained from the spiked sample of lowest concentration. The LODs of 16 PAHs and 15 NPAHs ranged from 0.018 to 1.7 and from 0.035 to 0.60 ng g−1, respectively. The LOQs were from 0.061 to 5.7 and 0.12 to 2.0 ng g−1 for 16 PAHs and 15 NPAHs, respectively. Table 1 presents the results obtained. The method showed a linear correlation between the peak area and analyte concentration over the full range of the calibration curve, with the coefficient of determination (R 2) ranging from 0.9901 to 0.9999 for all target compounds.
The precision of the method applied to standard solutions was studied within a day (n = 3, 50 and 5.0 × 102 ng g−1 standard solutions were injected by three needles within a day) and among days (n = 6, among 6 days, 50 and 5.0 × 102 ng g−1 standard solutions were injected by three needles each day. One group of data for each day was selected to obtain six groups of data for each standard solution to obtain the inter-day RSD) [26]. RSD values of the target compounds ranged from 0.10 to 5.7 and from 0.23 to 9.3% for the intra-day and inter-day RSD, respectively. It was proved that satisfactory precision was obtained for 16 PAHs and 15 NPAHs in this study.
Recovery and Precision
Recoveries and precisions were determined to confirm the application of the optimized method, and were measured by spiked ambient air particulate samples at three concentration levels (2.5 × 102, 5.0 × 102 and 1.0 × 103 ng g−1) for 16 PAHs and (1.0 × 102, 2.0 × 102, and 4.0 × 102 ng g−1) for 15 NPAHs before extraction, respectively. Table 2 summarizes the data of recoveries and precisions. Recoveries for 16 PAHs and 15 NPAHs ranged from 88.9 to 116 and from 81.6 to 111%, respectively. Good recovery results were achieved using the proposed method, compared to those from other reported methods (recoveries for PAHs and NPAHs ranged from 49.6 to 128 and from 60.0 to 104%, respectively) [6]. The precisions of the method for 16 PAHs and 15 NPAHs were in the ranges of 0.21–5.5% (n = 5) and 0.44–6.5% (n = 5), respectively, described as the RSD value. This indicates that the analytical method could be applied to the analysis of ambient air particulate samples.
Application to Real Samples
The proposed analytical method was applied to ten TSP samples collected in Beijing, China in June and December 2015. The quantitative results obtained are summarized in Electronic Supplementary Material Table S2, and show that average concentrations of 16 PAHs ranged from 10.90 to 18.97 ng m−3 in June and from 113.3 to 1213 ng m−3 in December. Average concentrations of 15 NPAHs ranged from 537.4 to 1239 pg m−3 in June and from 719.9 to 3726 pg m−3 in December. The concentrations of PAHs and NPAHs were remarkably higher in December than in June, and this data is in agreement with other reports from Beijing [20, 36]. During the summer and winter periods, phenanthrene, fluoranthene, pyrene, benzo(b)fluoranthene, benzo(a)anthracene, and chrysene were the prevalent PAHs, and 3-nitrophenanthrene and 3-nitrofluoranthene the predominant NPAHs, in TSP samples. In addition, Fig. 6 describes the concentration changes of 16 PAHs and 15 NPAHs during the June (summer) and December (winter) periods, and higher levels for both PAHs and NPAHs in December are clearly observed. In Fig. 6, polycyclic aromatic compounds (PAC) represent PAH and NPAH compounds. Particularly for PAHs, the percentage of four-ring PAHs was 57.4% in December samples, which were larger than that in June samples (27.0%). This could be attributed to increased emissions, mainly from fossil fuel usage for heating purposes, during the cold season. Because fossil fuels, such as coal, are widely used as the main fuels for heating purposes in Beijing, China [20, 36].

Concentration levels of 16 PAHs, 15 NPAHs at a sampling location in Beijing
Conclusion
A novel in-house built integrated ultrasonic extraction device, including an ultrasonic grinder instrument, extraction cell, and the purification tube, was proposed and successfully used for the extraction and purification of 16 PAHs and 15 NPAHs at low concentrations in ambient air particulates. In this study, the validated method showed good accuracy, satisfactory recoveries, and low LODs and LOQs in real samples. Furthermore, the method was applied successfully to evaluate the pollution level of these analytes in total suspended particulate samples in Beijing, China, in June and December 2015. A simple and effective analytical method was presented in this study. The application of which proved that it could fully meet the requirements for the determination of PAHs and NPAHs in ambient air particulates.
References
Shen H, Huang Y, Wang R, Zhu D, Li W, Shen G, Wang B, Zhang Y, Chen Y, Lu Y, Chen H, Li T, Sun K, Li B, Liu W, Liu J, Tao S (2013) Environ Sci Technol 47:6415–6424
Zhang Y, Tao S, Shen H, Ma J (2009) Proc Natl Acad Sci USA 106:21063–21067
Barrado AI, García S, Castrillejo Y, Barrado E (2013) Atmos Environ 67:385–393
Santos AG, Regis AC, da Rocha GO, Bezerra Mde A, de Jesus RM, de Andrade JB (2016) J Chromatogr A 1435:6–17
Yang XY, Igarashi K, Tang N, Lin JM, Wang W, Kameda T, Toriba A, Hayakawa K (2010) Mutat Res 695:29–34
Albinet A, Tomaz S, Lestremau F (2013) Sci Total Environ 450–451:31–38
Shamsipur M, Gholivand MB, Shamizadeh M, Hashemi P (2015) Chromatographia 78:795–803
Lung SC, Liu CH (2015) Sci Rep 5:12992
Qiao M, Qi W, Liu H, Qu J (2013) J Chromatogr A 1291:129–136
Cochran RE, Dongari N, Jeong H, Beranek J, Haddadi S, Shipp J, Kubatova A (2012) Anal Chim Acta 740:93–103
Albinet A, Nalin F, Tomaz S, Beaumont J, Lestremau F (2014) Anal Bioanal Chem 406:3131–3148
Masala S, Ahmed T, Bergvall C, Westerholm R (2011) Anal Bioanal Chem 401:3305–3315
Lin Y, Qiu X, Ma Y, Ma J, Zheng M, Shao M (2015) Environ Pollut 196:164–170
Yenisoy-Karakas S, Gaga EO (2013) Talanta 115:150–158
Walgraeve C, Chantara S, Sopajaree K, De Wispelaere P, Demeestere K, Van Langenhove H (2015) Atmos Environ 107:262–272
Mirivel G, Riffault V, Galloo JC (2010) Anal Bioanal Chem 397:243–256
Clark AE, Yoon S, Sheesley RJ, Usenko S (2015) Chemosphere 137:33–44
Han Y, Ren L, Xu K, Yang F, Li Y, Cheng T, Kang X, Xu C, Shi Q (2015) J Chromatogr A 1395:1–6
Di Vaio P, Cocozziello B, Corvino A, Fiorino F, Frecentese F, Magli E, Onorati G, Saccone I, Santagada V, Settimo G, Severino B, Perissutti E (2016) Atmos Environ 129:186–196
Li W, Wang C, Shen H, Su S, Shen G, Huang Y, Zhang Y, Chen Y, Chen H, Lin N, Zhuo S, Zhong Q, Wang X, Liu J, Li B, Liu W, Tao S (2015) Environ Pollut 197:156–164
Wang W, Jing L, Zhan J, Wang B, Zhang DP, Zhang HW, Wang DQ, Yang Y, Zhao J, Sun YF, Bi XH, Wang XT, Feng JL (2014) Atmos Environ 89:242–248
Albinet A, Leoz-Garziandia E, Budzinski H, Villenave E, Jaffrezo JL (2008) Atmos Environ 42:43–54
Portet-Koltalo F, Oukebdane K, Dionnet F, Desbene PL (2008) Anal Bioanal Chem 390:389–398
Pandey SK, Kim K-H, Brown RJC (2011) TrAC Trends Anal Chem 30:1716–1739
Géraud E, Prévot V, Leroux F (2006) J Phys Chem Solids 67:903–908
Huo F, Tang H, Wu X, Chen D, Zhao T, Liu P, Li L (2016) J Chromatogr B 1023–1024:44–54
Zhao T, Tang H, Chen D, Huo F, Li L (2017) Anal Methods 9:1168–1176
Wang CJ, Wu YA, Jacobs RMJ, Warner JH, Williams GR, O’Hare D (2011) Chem Mater 23:171–180
Bandowe BA, Meusel H, Huang RJ, Ho K, Cao J, Hoffmann T, Wilcke W (2014) Sci Total Environ 473–474:77–87
Wei C, Han Y, Bandowe BA, Cao J, Huang RJ, Ni H, Tian J, Wilcke W (2015) Sci Total Environ 505:814–822
Bandowe BAM, Wilcke W (2010) J Environ Qual 39:1349
Kanrar B, Mandal S, Bhattacharyya A (2010) J Chromatogr A 1217:1926–1933
Haubl G, Berthiller F, Krska R, Schuhmacher R (2006) Anal Bioanal Chem 384:692–696
Wang Y, Li S, Zhang F, Lu Y, Yang B, Zhang F, Liang X (2016) J Chromatogr A 1437:8–14
Wu J, Qian X, Yang Z, Zhang L (2010) J Chromatogr A 1217:1471–1475
Tang N, Suzuki G, Morisaki H, Tokuda T, Yang X, Zhao L, Lin J, Kameda T, Toriba A, Hayakawa K (2017) Atmos Environ 152:354–361
Acknowledgements
The study was funded by National Key R&D Program of China, No. 2016YFF0201102, the Basic Research of National Institute of Metrology, China, No.21-AKY1514 (Development of analytical method and certified reference materials for tracing air pollutant source), the National Natural Science Foundation of China (Grant no. 20773010), the Fundamental Research Funds for the Central Universities (Grant no. YS1406), and funded by the Undergraduate Innovation Program from Beijing University of Chemical Technology (Grant no. 2015-178305).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, C., Tang, H., Chen, D. et al. An Integrated Ultrasonic Extraction and Cleanup Procedure for Determining PAHs and NPAHs in Ambient Air Particulates. Chromatographia 80, 1433–1445 (2017). https://doi.org/10.1007/s10337-017-3360-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-017-3360-9