
Abstract
A stability-indicating reversed-phase high-performance liquid chromatography method using a photodiode array detector has been developed for simultaneous estimation of chlorthalidone (CLR) and azilsartan medoxomil (AZL) in combined solid dosage form. The method was developed based on statistical design of experiments (DoE) followed by optimization using the response surface methodology. Separation was achieved on a double end-capped C18 column (150 mm × 4 mm, 5 µm). The effects of % acetonitrile (v/v) and buffer salt concentrations on the retention time of the two drugs and on their resolution were investigated and optimized. A robust design space was created by the overlay contour plot method. The optimum chromatographic condition within the design space was found to be isocratic mobile phase consisting of 10 mM Tris(hydroxymethyl)aminomethane buffer (pH 7.7) and acetonitrile at ratio of 60:40 (v/v) with flow rate of 1 mL min−1 for 7 min. The retention times of CLR and AZL were found to be 2.6 and 4.9 min, respectively. The method was validated according to International Conference on Harmonization (ICH) and Food and Drug Administration (FDA) guidelines, and various validation parameters were determined. Forced degradation studies were also carried out in acid, base, oxidation, and reduction media with a view to establishing the specificity and stability-indicating property. The practical applicability of the method was confirmed by determining CLR and AZL in combined dosage form. This reliable and validated stability-indicating method for simultaneous estimation of CLR and AZL is available for routine analysis in the pharmaceutical industry as well as research laboratories.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Azilsartan medoxomil (AZL) is an angiotensin II receptor blocker used in adult patients with essential hypertension. Azilsartan medoxomil exhibits antihypertensive effect by selectively inhibiting binding of angiotensin II to angiotensin type 1 receptor, thereby antagonizing the pressor response activity of angiotensin II [1, 2]. In vitro, azilsartan produced greater and more sustained AT(1) receptor binding/blockade activity than several comparator angiotensin II receptor antagonists. Chlorthalidone (CLR), on the other hand, is considered to be a thiazide-like diuretic. Chlorthalidone has the longest duration of action but similar diuretic effect at maximal therapeutic doses. It is often used in management of hypertension and edema. Combination of these two drugs was proved superior to other similar classes of drugs and/or drug combinations for controlling primary blood pressure [3,4,5,6,7].
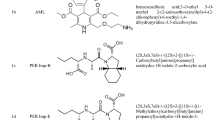
Chemically, AZL is (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl-2-ethoxy-1-{[2′-(5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl}-1H-benzimidazole-7-carboxylate [8] and CLR is 2-chloro-5-(1-hydroxy-3-oxo-2,3-dihydro-1H-isoindol-1-yl)benzene-1-sulfonamide [9]. The chemical structures are presented in Fig. 1.

Chemical structure of a azilsartan medoxomil and b chlorthalidone
A number of high-performance liquid chromatography (HPLC) methods have been reported for simultaneous estimation of these two drugs in combined dosage form [10,11,12,13], but none of them followed a systemic approach for method development. In the presented research, a method was developed and optimized using design of experiments (DoE) and the optimum condition picked from the design space. Thorough validation work was also carried out according to ICH, United States Pharmacopeia (USP), and FDA guidelines.
Materials and Methods
Materials
Working standards were a kind gift from ACI Pharmaceuticals Limited, Bangladesh. HPLC-grade acetonitrile and methanol were obtained from Active Fine Chemicals Ltd., Bangladesh.
Diluting Solvent
AZL is reported to be relatively stable in the pH range from 3 to 5 [14]. Consequently, in the current research, a blend of 10 mM acetate buffer solution (pH 4.0) and acetonitrile at ratio of 60:40 was used as diluting solvent.
Standard Solution Preparation
Working standards of 25 mg CLR and 40 mg AZL were taken in a 100-mL volumetric flask, and 5 mL methanol was added to wet the mass, followed by filling to the mark with diluting solvent to obtain stock standard solution. Suitable serial dilutions were carried out to obtain the nominal concentration (12.5 μg mL−1 CLR and 20 μg mL−1 AZL) and another five concentrations from 50 to 200% of nominal.
Sample Solution Preparation
For assay preparation, 20 tablets (Edarbyclor 40 mg/25 mg) were ground, and tablet powder equivalent to 25 mg CLR and 40 mg AZL was taken in a 100-mL volumetric flask and suitably diluted with mobile phase.
Chromatographic Condition
A double end-capped C18 column (150 mm × 4 mm, 5 µm) was used for this study. All analyses were carried out at ambient temperature and at the flow rate of 1 mL min−1. The injection volume was 20 µL for standard and samples. Before injection, each standard and sample was filtered through 0.2-µm filter tips. Column eluents were monitored using a photodiode array detector (PDA) at 249 nm.
Design for Optimization
Experimental design using the response surface methodology (RSM) was applied for optimization of the mobile phase of the proposed method. A central composite design (CCD) with two factors and a total of 13 runs was selected for the optimization study.
The independent variables and their levels were selected based on previous knowledge and preliminary screening. The dependent variables (responses) were the retention time of the two drugs and the resolution between them. The design matrix and the obtained responses were subjected to multiple regression analysis, and the resulting second-order polynomial function was used to correlate the independent variables and the responses. Variables and their limits and responses with specific targets are presented in Table 1.
Statistical Analysis
Experimental design and statistical analysis were carried out using Design-Expert 10.0.2 (free trial version). The significance of each model, terms, and their interactions were tested by analysis of variance (ANOVA). Any term or model having p value less than 0.05 was considered statistically significant. Also, F values for each model and term were checked to assess the significance of experiments. F value of 1 or close to 1 indicates that the null hypothesis is true (i.e., that experimental factors have no effect on responses), while large F value indicates that the effect of factors on responses is significant. The fit of the data by the regression line was assessed based on the determination coefficient (R 2), adjusted determination coefficient (adj. R 2), and predicted determination coefficient (pred. R 2). Model adequacy was checked using diagnostic tools such as the normal probability plot of residuals and residual versus predicted plot.
Validation Parameters
The developed method was validated according to ICH, USP, and FDA guidelines with respect to specificity, solution stability, linearity and range, accuracy, precision, system suitability, and forced degradation study.
Specificity
The specificity of the LC method was evaluated to ensure that there was no interference from degradation products, excipients, or other impurities with the drug peaks in the chromatogram. Visual inspection as well as peak purity indices were used to evaluate the specificity of the drugs in the chromatogram.
Solution Stability
The stability of the drug combination in the diluting solvent was checked by keeping test solutions in tightly capped vials at room temperature and in refrigerator at 5 °C for 48 h. The solutions were analyzed using the developed method at 0, 24, and 48 h. Area changes were compared with the initial state.
Linearity and Range
To evaluate the linearity and range of the method, a calibration curve was constructed by plotting peak area against various concentrations (50–200% of nominal). The linear equation, regression coefficient (r 2), intercept, and slope were calculated.
Accuracy
The ICH recommends collecting data from a minimum of nine determinations over a minimum of three concentration levels covering the specified range. In the present study, accuracy was determined based on the % recovery at five different concentrations.
Precision
Precision is usually assessed at two levels, namely intraday (repeatability) and interday (intermediate precision). In the current study, intraday precision was assessed at three concentration levels using three replicates at each level. Intermediate precision was assessed using two HPLC systems on two different days. The % relative standard deviation (RSD) of the total variation was calculated.
System Suitability
System suitability is commonly used to ensure the adequacy of a method for a particular analysis. The following parameters were verified in the current study: theoretical plate count, tailing factor, resolution and reproducibility of area, limit of detection (LOD), and limit of quantification (LOQ).
Forced Degradation Study
Forced degradation studies are undertaken to degrade a sample (e.g., drug product or drug substance) deliberately. These studies are used to evaluate the specificity of an analytical method. Forced degradation experiments also provide important information on a drug’s degradation pathway, in turn helping rational formulation development of that drug in different dosage forms. Drug substances or drug products are exposed to acid, base, oxidizing agent, reducing agent, and water to produce 10–30% degradation of the drug substance. The degraded samples are then analyzed using the method to determine whether there is any interference between the drug molecule and related compound(s).
Results and Discussion
Method Development
Preliminary Screening
To determine the significant factors and their effects and levels, preliminary screening and literature search were carried out. Screening experiments revealed that acetonitrile in the mobile phase in the range from 30 and 50% (v/v) could separate CLR and AZL with acceptable resolution, whereas beyond 50% acetonitrile the peaks merged. On the other hand, methanol alone with buffer solution in mobile phase resulted in longer retention of the drug molecules. Different buffer systems and pH values were investigated. As a rule of thumb, the pH of the mobile phase buffer should be 2 units above or below the pK a value of the drugs [15, 16]. The pK a values of AZL and CLR are 6.1 and 9.4, respectively. At pH of 2 units below 6.1 (i.e., ≤4.1), AZL remains undissociated, resulting in longer retention and requiring larger % acetonitrile to reduce its retention. Therefore, we selected pH 7.7, which is almost two units above the pK a value of AZL but two units below the pK a value of CLR, indicating that both drugs should remain dissociated at this pH. As mobile phase buffer, there were two options: phosphate buffer and Tris(hydroxymethyl)aminomethane buffer. In the current case, Tris(hydroxymethyl)aminomethane buffer was chosen, due to the fact that the larger molecular structure of the Tris salt is less harmful to the column when the mobile phase pH is near to or above 8.0 [17]. Again it was revealed that the Tris buffer concentration had a profound effect on the retention of AZL. Therefore, we chose the acetonitrile and Tris buffer concentrations as the two independent variables for optimization. Other factors such as temperature, flow rate, etc. were fixed based on knowledge and experience. Separation was carried out on a double end-capped C18 column (150 mm × 4 mm, 5 µm) at ambient temperature. The flow rate was set at 1.0 mL min−1 with injection volume of 20 µL. Eluents were monitored at 249 nm.
Experiments and Statistical Analysis
A full factorial design with 13 experimental conditions was performed, including nine conditions defined by the design of experiments with five repetitions at the center of the domain. A chromatogram was recorded for each of these conditions. The runs and corresponding responses are depicted in Table 2. The retention of CLR varied from 1.90 to 4.05 min, while that of AZL varied from 2.22 to 15.10 min. These data could therefore clearly be utilized and analyzed to determine the optimum condition to achieve the target of the current study.
ANOVA was conducted to test the significance of the experimental quadratic models, the results of which are presented in Table 3. The significance of each model was assessed based on Fisher’s ratio (F value). The F values for the CLR model, AZL model, and R s model were found to be 1765.30, 24,686.77, and 827.48, respectively. The p values for all three models were found to be less than 0.0001, clearly indicating that the models were significant. All the other terms were also significant, as is evident from their corresponding F and p values. The quality of the obtained polynomial regressions was assessed based on the determination coefficient (R 2), adjusted determination coefficient (adj. R 2), and predicted determination coefficient (pred. R 2). In all cases, the R 2 values were found to be quite close to 1, indicating data fit by the regression curve with accuracy of more than 99%. The predicted R 2 values were in reasonable agreement with the adjusted R 2 values (i.e., difference less than 0.2). Adequate precision can be assessed based on the signal-to-noise ratio, for which a value greater than 4 is desirable [18]. In the current study, high values of adequate precision were found, indicating an adequate signal. Table 4 summarizes the model. The polynomial equations in coded form for each model were as follows:
Here, A is the ACN % v/v (acetonitrile) and B is the salt concentration (mM). A and B represent the main effect terms, while AB represents their interaction effect.
To check the adequacy of the model, visual inspection of the normal probability plot and residual versus predicted plot was carried out. It was observed that residuals were normally distributed along the straight line with minor scatter, indicating good fit to the data. The residual plots showed random distribution of residuals within +4 and −4 without any trend, indicating absence of any systematic bias or outliers. The graphs are presented in Fig. 2.

Normal probability plot and residual plot of a CLR model, b AZL model, and c R s model
Design Space and Optimal Separation Condition
The standardized effects of the independent factors and their interactions on the responses were visualized using three-dimensional response surface graphs and two-dimensional overlay contour plots (Fig. 3). Using the data for the experimental conditions and corresponding responses, an overlay contour plot was constructed to determine the design space and the optimal composition of mobile phase. In this case, the target was to minimize the retention time of both drugs while simultaneously maximizing the resolution. More precisely, the target was to achieve minimum and maximum responses of 2.5 ≤ R t of CLR ≥3.5, 4 ≤ R t of AZL ≥6, and R s ≥ 5. The desirability of different conditions within the design space was considered, choosing the most favorable. Mobile phase with around 40% (v/v) acetonitrile and buffer concentration of around 10 mM met the entire target with desirability value of 1.00. Finally, the optimum isocratic mobile phase was chosen as a blend of acetonitrile 40% (v/v) and 10 mM Tris(hydroxymethyl)aminomethane buffer 60% (v/v).

Three-dimensional (3D) graphs of interaction effect of factors on a retention time of CLR, b retention time of AZL, and c resolution (R s); d overlay contour plot of interaction effects of factors on responses. Green zone indicates the design space of the method
Method Validation
Specificity
According to visual inspection of the standard chromatogram, assay sample chromatogram, and forced degradation sample chromatogram, no interference by any other peak of impurities or excipients was found. Moreover, peak purity indices in all cases were checked and found to be above 0.9998, indicating specificity of the method for the drug molecules. Figure 4a shows the chromatograms of standard mixture and sample mixture. Figure 4b shows the peak purity detected by the PDA detector.

a Chromatogram of standard mixture and sample mixture; b peak purity of CLR and AZL
Solution Stability
To check stability in this solvent, vials containing working standard at nominal concentration were kept at room temperature and in refrigerator, and tested at 0, 24, and 48 h. Then, the peak areas were compared with that at 0 h. No significant change in area was observed. The % RSD was found to be below 2.0%, indicating stability of the drugs in the diluting solvent.
Linearity, Working Range, and Accuracy
In the current validation procedure, the nominal standard concentration was 12.5 µg mL−1 CLR and 20 µg mL−1 AZL. We prepared 50, 80, 120, and 200% of the nominal standard solution and plotted the peak areas against the corresponding concentration. The determination coefficient (r 2) was found to be above 0.999 for both drugs, indicating acceptable fit of the data by the regression line. The working range was derived from the linearity and found to be 6.25–25 µg mL−1 for CLR and 10–40 µg mL−1 for AZL. The regression equations were as follows:
Percent recoveries were well within the limit (100 ± 2%), indicating accuracy of the method for simultaneous estimation of the drugs. The results are presented in Table 5.
Precision
In the present study, three different solutions were prepared with known added amounts of drugs (50, 100, and 200% of nominal concentration) and injected in triplicate. Percent recoveries (concentrations) were calculated by regression equation. The results of the precision study are presented in Table 6. The low % RSD of the total variation clearly indicates that the method is precise within the desired recovery range.
System Suitability
All the system suitability parameters met the desired level. The results are presented in Table 7.
Forced Degradation Study
In the current work, forced degradation studies were carried out in 0.1 N HCl solution, 0.1 N NaOH solution, 10% H2O2 solution, and 10% sodium metabisulfite solution. Samples were kept in the dark for 24 h, then analyzed using the newly developed method. Peak area was compared with that of freshly prepared standard solution. It was observed that AZL was significantly sensitive to all conditions except acidic media; It showed complete degradation in basic environment and in reduction condition. AZL was also highly sensitive to acidic media. CLR, on the other hand, showed reasonable stability in all conditions, showing the greatest degradation in acidic media. This information can be utilized for development of AZL and CLR combined tablet formulations. As AZL showed significant degradation propensity in acidic and basic media but was reasonably stable in diluting solvent (pH 4.0), buffering agent can be incorporated into tablet granules to maintain pH of around 4.0. The results are presented in Table 8. Degradation chromatograms are shown in Fig. 5.

Chromatograms of forced degradation samples for acid, base, reduction, and oxidation (clockwise from top-left corner)
Practical Applicability of Method
Edarbyclor 40/12.5 and Edarbyclor 40/25 were collected and assayed using the method. Recoveries were found to be within 100 ± 2% for both CLR and AZL.
Conclusions
Development of rapid, reliable, and easy-to-use analytical methods is a prime concern in the pharmaceutical industry to ensure the quality of tablet dosage forms in terms of assay, content uniformity, and stability during formulation and for quality control. In this work, a rapid, simple, and stability-indicating method was developed for simultaneous estimation of CLR and AZL in solid dosage form. Design of experiments was successfully applied in the development of this method, and a robust design space determined. The method was validated based on the validation guidelines for analytical methods outlined by the ICH, FDA, and USP. Forced degradation studies were carried out under various conditions, providing important information for development of combined solid dosage forms of these two drugs. Hence, the results of this study will be very useful to researchers as well as industrial personnel.
References
Perry CM (2012) Azilsartan medoxomil: a review of its use in hypertension. Clin Drug Invest 32(9):621–639. doi:10.2165/11209600-000000000-00000
Zaiken K, Cheng JW (2011) Azilsartan medoxomil: a new angiotensin receptor blocker. Clin Ther 33(11):1577–1589. doi:10.1016/j.clinthera.2011.10.007 (Epub 2011 Nov 8)
Cheng JW (2013) Azilsartan/chlorthalidone combination therapy for blood pressure control. Integr Blood Press Control 6:39–48. doi:10.2147/IBPC.S34792
Cushman WC, Bakris GL, White WB, Weber MA, Sica D, Roberts A, Lloyd E, Kupfer S (2012) Azilsartan medoxomil plus chlorthalidone reduces blood pressure more effectively than olmesartan plus hydrochlorothiazide in stage 2 systolic hypertension. Hypertension 60(2):310–318. doi:10.1161/HYPERTENSIONAHA.111.188284 (Epub 18 Jun 2012)
Pierini D, Anderson KV (2013) Azilsartan medoxomil/chlorthalidone: a new fixed-dose combination antihypertensive. Ann Pharmacother 7(5):694–703. doi:10.1345/aph.1R618 (Epub 12 Apr 2013)
Baker WL, Nigro SC, White WB (2014) Efficacy of azilsartan medoxomil with chlorthalidone in hypertension. Expert Rev Cardiovasc Ther 12(7):791–798. doi:10.1586/14779072.2014.924853 (Epub 2 Jun 2014)
Shuster JE, Bleske BE, Dorsch MP (2012) Clinical utility of azilsartan-chlorthalidone fixed combination in the management of hypertension. Vasc Health Risk Manag 8(1):381–387. doi:10.2147/VHRM.S22583
https://pubchem.ncbi.nlm.nih.gov/compound/azilsartan_medoxomil#section=3D-Conformer. Accessed 14 June 2016
https://pubchem.ncbi.nlm.nih.gov/compound/chlorthalidone#section=Names-and-Identifiers. Accessed 14 June 2016
Sravani P, Kumar SR, Duganath N, Devanna N (2014) Method development and validation for the simultaneous estimation of azilsartan and chlorthalidone by RP-HPLC in pharmaceutical dosage form. Int J Pharma Sci 4(5):725–729
Naazneen S, Sridevi A (2014) Stability-indicating RP-HPLC method for the simultaneous estimation of azilsartan medoxomil and chlorthalidone in solid dosage forms. Int J Pharm Pharm Sci 6(6):236–243
Sohni SK, Kumar R, Akhtar M, Ranjan C, Chawla G (2016) Development and validation of RP-HPLC method for simultaneous estimation of azilsartan medoximil and chlorthalidone in bulk form and formulation using quality by design. Int J Pharm Pharm Sci 8(2):266–272
Kasimala MB, Kasimala BB (2012) Reverse phase HPLC method development and validation for the simultaneous estimation of azilsartan medoxomil and chlorthalidone in pharmaceutical dosage forms. J Atoms Mol 2(1):117–126
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/200796orig1s000chemr.pdf. Accessed 22 December 2016
Ahuja S, Scypinski S (2001) Handbook of modern pharmaceutical analysis, 1st edn, vol III. Academic, San Diego, p 353
Ohannesian L, Streeter AJ (2002) Handbook of pharmaceutical analysis. Marcel Dekker, New York, p 233
Kayesh R, Sultan MZ (2015) A novel ion-pair RP-HPLC method for simultaneous quantification of naproxen and esomeprazole in pharmaceutical formulations. J Chromatogr Sci 53(5):687–693. doi:10.1093/chromsci/bmu103
Montgomery DC (2004) Design and analysis of experiments, 5th edn, pp 104–107
Acknowledgements
The authors are very grateful to the Director of Quality Assurance of ACI Limited for providing the active ingredients and reagents.
Author contribution
The idea was developed by R.K. Preliminary screening, design of experiments, and statistical analyses were performed by R.K. M.S.J. prepared the validation protocol and performed the validation and forced degradation study for the method. R.K. wrote the first draft of the manuscript and arranged the references. The whole research was supervised and supported by M.Z.S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding/grant was received for conducting this study.
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Kayesh, R., Jahan, M.S. & Sultan, M.Z. Development Using Response Surface Methodology and Validation of a Stability-Indicating RP-HPLC Method for Simultaneous Estimation of Azilsartan Medoxomil and Chlorthalidone in Solid Dosage Form. Chromatographia 80, 593–603 (2017). https://doi.org/10.1007/s10337-017-3276-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-017-3276-4