
Abstract
This work reports the development and validation of an ultra-high-performance liquid chromatography method to separate 11 different cardiovascular drugs: acenocoumarol, amiloride, bisoprolol, fluvastatin, furosemide, glibenclamide, hydrochlorothiazide, rosiglitazone (internal standard), valsartan, verapamil and warfarin. Detection of the analytes was carried out by means of photodiode array and fluorescence detection. Initially, the most significant variables in liquid chromatography (pH of the mobile phase, nature of the organic modifier and stationary phase type) were studied simultaneously in a systematic way. Taking into account the resolution obtained for the different chromatographic separations, pH 4.5, methanol and C18 were chosen as mobile phase pH, organic modifier and stationary phase, respectively. The method was validated and applied to the analysis of real human plasma samples using a solid phase extraction sample treatment, and the robustness of the chromatographic separation was studied by experimental design. The reported method allows the separation of all the analytes and the IS in less than 6 min, and, furthermore, it simplifies the analysis of drugs used in combined cardiovascular therapy by applying a fast and simple methodology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It is estimated that noncommunicable diseases (NCDs) kill approximately 38 million people each year [1]. The main types of NCDs, also known as chronic diseases, are cardiovascular diseases (CVDs), cancers, chronic respiratory diseases and diabetes. Among them, CVDs cause around 17.3 million deaths per year and this number is expected to be more than 23.6 million by 2030 according to current estimation [2].
Several risk factors, as hyperlipemia, arterial hypertension, obesity or diabetes mellitus, are associated with CVDs. These factors act synergically, increasing the occurrence of CVDs. The treatment of several CVDs may include lifestyle changes as well as pharmacological therapy using drugs that have different targets and mechanisms of action to address the risk factors. The drugs used in cardiovascular pharmacotherapy are antihypertensives (including different families of diuretics, vasodilators, cardioinhibitors and central sympatholytic drugs), hypolipidemic agents, anticoagulants, antiplatelets, and hypoglucemic drugs. In order to tackle the different risk factors, two or more drugs of these different families are commonly simultaneously prescribed, what is known as combined cardiovascular therapy. Some situations that may hamper the success of the therapeutic treatment are inadequate prescription or dosing of medication, interactions between prescribed drugs, or poor adherence to treatment. With the aim of increasing the adherence to treatment of the patients by simplifying the treatment regimens and reducing costs, fixed-dose combination medicines (polypills) have been developed combining in a single pill several drugs used in cardiovascular therapy [3, 4].
The common combination of drugs for the treatment or prevention of CVDs implies the necessity to develop methods able to determine simultaneously several drugs from different families in complex matrices such as biological fluids to be applied in pharmacokinetic studies, efficacy and toxicity assessment, medication adherence control or doping analysis. These methods should be simple, fast, and easy to optimize, so that they can be rapidly implemented in routine analysis when necessary.
Liquid chromatography is one of the main analytical techniques nowadays used in the pharmaceutical industry. Numerous methods based on high-performance liquid chromatography (HPLC) have been developed for the analysis of cardiovascular drugs [5,6,7,8]; however, the shorter analysis times, less consumption of solvents and better resolution and sensitivity of ultra-high-performance liquid chromatography (UHPLC) systems have caused an increase in the use of this technique [9,10,11].
Mass spectrometry (MS) detection is commonly the first choice for the analysis of complex mixtures of drugs [12,13,14], as it combines optimum sensitivity and great identification capability. However, the photometric and fluorimetric characteristics of cardiovascular drugs make them excellent candidates for photometric and fluorescence detection.
The development of suitable analytical methods is indeed one of the most challenging aspects in pharmaceutical analysis. Although several approaches based on experimental design have been proposed to overcome the problems derived from one variable at a time (OVAT) optimization procedures [15,16,17], the success of these alternatives is sill only moderate. In this work, a systematic approach is applied to the optimization of a method using an UHPLC coupled to photodiode-array (PDA) and fluorescence (FLD) detectors for the determination of 11 cardiovascular drugs in plasma: acenocoumarol (anticoagulant agent), amiloride (potassium-sparing diuretic), bisoprolol (β-blocker), fluvastatin (lipid-lowering drug), furosemide (loop diuretic), glibenclamide (sulfonyluric antidiabetic drug), hydrochlorothiazide (thiazidic diuretic), rosiglitazone (thiazolidinedionic antidiabetic drug), valsartan (ARA-II), verapamil (calcium channel blocker) and warfarin (anticoagulant agent).
Plasma samples were treated with a protein precipitation with acetonitrile followed by a solid phase extraction process using strong cation exchange mixed mode cartridges. The optimized method was validated in terms of recovery, selectivity, calibration range, accuracy and precision, and applied to the analysis of real samples. In addition, the robustness of the method was studied using experimental design.
Materials and Methods
Instrumentation
Analyses were performed on an Acquity UPLC system (Waters, Milford, USA), coupled to a PDA detector and a FLD detector. The PDA detector collected spectra from 190 to 400 nm with a data sampling rate set to 20 points s−1. The FLD detector worked with two different channels at the selected optimum excitation and emission wavelengths with a sampling rate of 2 points s−1. System control, data collection and data processing were accomplished using Empower 2 software (Waters). The chromatographic columns used to perform the separation were a BEH C18, 1.7 μm, 50 × 2.1 mm (Waters) and a Kinetex PFP 1.7 μm, 50 × 2.1 mm (Phenomenex, CA, USA). Strong cation exchange mixed mode Strata X-C cartridges (Phenomenex) (30 mg/1 mL) were used for the sample treatment of plasma. The pH of buffer solutions was measured with a Crison GPL 22 pH-meter (Barcelona, Spain) equipped with a Crison 5209 electrode.
Chemical and Reagents
Acenocoumarol, amiloride hydrochloride, furosemide, rosiglitazone maleate (internal standard, IS), verapamil hydrochloride and warfarin were kindly provided by the Institute of Forensic Medicine in Freiburg (Germany). Sodium fluvastatin and valsartan were supplied by Novartis (Basel, Switzerland) and bisoprolol by Merck (Darmstad, Germany). Hydrochlorothiazide and glibenclamide were purchased from Sigma Aldrich (MO, USA). The 1000-mg L−1 stock solutions for each compound were prepared in methanol (MeOH) or acetonitrile (ACN) and stored at 4 °C.
Pro-analysis grade acetic acid, formic acid, ammonium acetate, ammonium formate, disodium hydrogen citrate sesquihydrate, trisodium citrate dehydrate, ammonium chloride and ammonium hydroxide from Sigma Aldrich were used in the preparation of the buffer solutions. Gradient quality MeOH (Romil, Cambridge, USA) and ACN (Baker, Deventer, Netherlands) were used as organic modifiers. Purified water from a Milli-Q Element A10 system (Millipore, MA, USA) was used in the preparation of buffer and reagent solutions.
Drug-free human plasma samples were purchased from the Blood Bank of Galdakao Hospital (Biscay, Basque Country) and collected in polypropylene tubes to be frozen at −20 °C.
Photometric and Fluorimetric Study: Influence of pH on the Absorption and Fluorescence Spectra
To study the photometric and fluorimetric properties of the analytes a 5-mg L−1 standard solution of each analyte in 25:75 buffer solution:MeOH (v:v) was injected individually into the chromatographic system. The effect of the pH in the absorption and fluorescence spectra was studied using four different buffer solutions ranging from pH 2.5 to 8.5 as aqueous mobile phases: pH 2.5 (formic/formate, 5 mM), pH 4.5 (acetic/acetate, 5 mM), pH 6.5 (citric/citrate, 5 mM) and pH 8.5 (ammonium/ammonia, 5 mM). MeOH was used as organic modifier. A linear gradient at a constant flow of 0.4 mL min−1 was used, starting from 10% of organic modifier to reach 95% in 3 min.
Absorption spectra were collected by the PDA detector from 190 to 400 nm. The optimum excitation and emission wavelengths for fluorimetric detection were obtained, first setting a tentative excitation wavelength (λ ex) from the absorption spectrum, and then acquiring the emission spectra in a wavelength range of 70 nm starting from the λ ex. Once the optimum emission wavelength (λ em) was found, the excitation spectrum was acquired and the optimum λ ex was selected.
Chromatographic Optimization
The most significant and most commonly optimized variables in a chromatographic separation are the pH of the mobile phase, the nature of the organic modifier and the stationary phase of the column [18]. Correspondingly, mobile phases buffered at three different pH values, two organic modifiers, and two chromatographic columns were tested in a systematic way in order to find the best separation method for the 11 cardiovascular drugs studied.
As in the photometric and fluorimetric study, it was observed that the absorption and fluorescent properties of some of the studies analytes depended on the pH, the working range was limited between 3.5 and 5.5, and the aqueous mobile phase pH values studied were 3.5, 4.5 and 5.5. An amount of 10% of the corresponding organic modifier was added to all the aqueous mobile phases in order to avoid bacterial growth. The organic modifiers tested as organic mobile phases were the most widely used in liquid chromatography MeOH and ACN. Finally, the influence of the stationary phase was studied using two columns: a C18 BEH column (one of the most common chromatographic columns used in the analysis of cardiovascular drugs) [10] with a retention mechanism based on hydrophobic interactions, and a PFP (pentafluorophenyl) column that, in addition to the hydrophobic interactions, includes other retention mechanisms (aromatic interaction, dipole–dipole interaction and hydrogen bonding).
All these variables were systematically studied carrying out the 12 possible combinations of experiments. For this aim, 5 µL of a standard solution in 75:25 aqueous mobile phase:MeOH (v:v) containing 5 mg L−1 of each analyte was repeatedly injected. Two different gradients were applied depending on the organic modifier: from 10 to 95% of organic mobile phase in 5 min when MeOH was used, and from 10 to 90% in 5 min when using ACN, in order to compensate the higher eluotropic strength of this solvent. The flow was kept at 0.4 mL min−1 and the highest proportion of organic solvent was held for 1 min before returning to the initial conditions for column cleaning purposes.
After carrying out these experiments, the most suitable combination of organic modifier, stationary phase and pH was chosen, using as selection criteria the peak resolution of the adjacent chromatographic peaks.
Once the optimum values for the most significant variables were fixed, the gradient was segmented in 2-min steps and the slope was optimized in each step to maximize the separation. Moreover, the best reconstitution solution and injection volume were studied. Standard samples containing 1 µg of each compound were evaporated to dryness under a stream of N2 and reconstituted to 200 µL with proportions of aqueous mobile phase:MeOH (v:v) ranging from 100:0 to 0:100. To optimize the injection volume, 2.5, 5.0 and 7.5 µL were tested.
Sample Treatment
A previously optimized solid phase extraction (SPE) process was used for the treatment of plasma samples. An amount of 500 μL of human plasma sample spiked with 25 μL of a 20-mg L−1 rosiglitazone methanolic solution as IS was transferred to a 2-mL Eppendorf tube. To promote protein precipitation, 725 μL of acetonitrile were added to the sample. After vortex mixing and centrifugation for 5 min at 10,000 rpm and 20 °C, the supernatant was transferred to a 6-mL glass tube and the excess or organic solvent was evaporated at 60 °C under a stream of N2 for 5 min. Next, 500 μL of a 0.1% formic acid solution were added to the resulting solution, which was vortex-mixed and transferred to a strong cation mixed mode Strata X-C cartridge, previously activated and conditioned with 1 mL of MeOH and 1 mL of a 0.1% formic acid solution, respectively. After the addition of the sample, the cartridge was washed with 500 μL of a 0.1% formic acid solution containing 3.4% of MeOH. The analytes were eluted using 2.5 mL of a methanolic solution of ammonium hydroxide (2.2%). The eluent was collected in a glass tube and evaporated to dryness at 60 °C under a stream of N2. The residue was reconstituted with 500 μL of a solution of aqueous phase:methanol (55:45), vortex mixed, filtered with GHP filters (hydrophilic polypropylene, 0.2 µm, 13 mm Ø) supplied by PALL (Ann Arbor, MI, USA), transferred to chromatographic vials and injected into the UHPLC system for analysis.
Validation
The recovery, calibration range, accuracy, precision and selectivity of the method were validated following an in-house validation protocol based on FDA and EMA guidelines [19, 20]. Moreover, the robustness of the chromatographic separation was thoroughly studied by means of experimental design.
Recovery, Calibration Range, Accuracy, Precision, and Selectivity
To assess the recovery, blank plasma samples were spiked with the analytes at 1 mg L−1 concentration before and after the extraction by SPE. Recovery was calculated as the ratio of the response for the analyte in the sample prior to extraction (n = 5) and after the extraction (n = 5).
Calibration curves for the different analytes were built in the range 0.04–5 mg L−1 (n = 9) using multi-component standard solutions by plotting the peak area of each analyte divided by the area of the IS (A analyte/A IS) against its nominal concentration. Calibration standards were prepared in drug-free human plasma and were analyzed in triplicate for each concentration level on 3 different days. Their concentration value was obtained from interpolation of the resulting corrected area in the regression equation of the calibration curve.
Accuracy was expressed as relative error (%RE) calculated as the deviation of the mean concentration value obtained from the replicates of each calibration point from their nominal concentration value. Intra- and inter-day precision was expressed as relative standard deviation (%RSD) among replicates. The lowest concentration calibration standard that fulfilled the accuracy and precision criteria of %RE and %RSD <20% was considered to be the lowest limit of quantitation (LLOQ) and the first point of the calibration range. For the rest of calibration points, the acceptance criterion for accuracy and precision was %RE and %RSD <15%.
Selectivity of the method was evaluated by comparing the responses of six blank plasma samples obtained from healthy volunteers against a sample at the LLOQ, with reference to potential endogenous and environmental interferences. To consider the method selective, the signal in the blank plasma must be lower than 20% the response of the analytes under study and in the case of the IS lower than 5% of the area obtained at the concentration used.
Robustness of the Chromatographic Separation
Experimental design was used to study the effect of slight variations of the studied variables that may have a significant influence on the separation: pH of the aqueous mobile phase, temperature of the column, flow rate, reconstitution solution composition and percentage of organic modifier in aqueous mobile phase. For this purpose, a fractional factorial design (resolution V) was employed using The Unscrambler software (CAMO, Oslo, Norway) for data treatment [21].
The uncertainty given by the pH meter, column oven, pumping system and volumetric material were used to establish the range of variation of the studied variables [22,23,24]. Applying the coefficient for extreme levels (k) to the calculated uncertainty, the whole expected range of variation was covered. The coefficients for extreme levels and the working ranges are gathered in Table 1.
Peak resolution of the most critical separations—verapamil-rosiglitazone, acenocoumarol-valsartan and valsartan-warfarin—were used as response variables. Also, peak width of the early eluting analytes hydrochlorothiazide and amiloride, and peak area of more non polar analytes glibenclamide and fluvastatin, were included as response variables, as variations in the reconstitution solution composition may affect the peak shape of the early eluting analytes and the dissolution of the most non-polar analytes.
An eight-experiment (25−2) fractional factorial design with three central samples was built. The responses obtained from these experiments were statistically treated by analysis of variance (ANOVA) to check which of the studied variables have a significant effect.
Analysis of Real Samples
Blood samples were collected from different patients under treatment with at least one of the analyzed drugs or a combination of them. These patients were also treated with other co-administered drugs. Blood samples were immediately transferred into tubes containing EDTA. Then, they were centrifuged at 1.301g for 10 min at 4 °C. The plasma supernatant was carefully separated from blood cells and collected in polypropylene tubes to be frozen at −20 °C until analysis. The analysis was carried out as explained in “Sample Treatment”.
Results
Photometric and Fluorimetric Study
Among the studied compounds, none of them showed a significant absorption dependence on pH. However, it was observed that the fluorescence of some analytes depended on pH: furosemide and valsartan were not fluorescent at either pH 6.5 or 8.5, while rosiglitazone was not fluorescent at pH 2.5. Therefore, the working range for the mobile phase was set between pH 3.5 and 5.5. Acenocoumarol and hydrochlorothiazide were not fluorescent at any pH value; thus, they can be only analyzed photometrically. For glibenclamide, the fluorescence observed was very weak, the PDA detection being more sensitive.
The optimum λ ex and λ em obtained for fluorimetric analysis together with the absorption maxima wavelengths for photometric analysis are collected in Table 2.
Chromatographic Optimization
The chromatograms obtained with the 12 possible combinations of experiments are gathered in Figs. 1 and 2. Using the PFP column (Fig. 1), both verapamil and bisoprolol were strongly retained, and, when using MeOH as organic modifier, none of these analytes eluted from the column in the time of the analysis. When ACN was used, verapamil eluted extremely late when using pH 5.5 and pH 3.5, and it did not elute at pH 4.5. Also, bisoprolol eluted at late times of analysis and, in addition, its chromatographic peak shape was broad and asymmetric.

Chromatograms obtained with the PFP column at different pH values using a methanol or b acetonitrile as organic modifier for a 5-mg L−1 standard solution containing hydrochlorothiazide (1), amiloride (2), furosemide (3), bisoprolol (4), rosiglitazone (5) verapamil (6), acenocoumarol (7), valsartan (8), warfarin (9), glibenclamide (10) and fluvastatin (11) at the working wavelength of 230 nm

Chromatograms obtained with BEH C18 column at different pH values using a methanol or b acetonitrile as organic modifier for a 5-mg L−1 standard solution containing hydrochlorothiazide (1), amiloride (2), furosemide (3), bisoprolol (4), rosiglitazone (5) verapamil (6), acenocoumarol (7), valsartan (8), warfarin (9), glibenclamide (10) and fluvastatin (11) at the working wavelength of 230 nm
Contrastingly, the BEH C18 column (Fig. 2) was able to properly retain all the compounds of interest in the proposed time of analysis; thus, it was chosen as the appropriate column for this chromatographic analysis.
Observing all the separations obtained using the BEH C18 column, those performed with MeOH as organic modifier showed better resolution than the ones obtained using ACN. Among them, pH 4.5 is the only pH value at which none of the compounds coelute. Thus, the best chromatographic separation obtained, in terms of resolution, was the one corresponding to the BEH C18 column, MeOH as organic modifier, and pH 4.5 buffer solution as aqueous mobile phase.
The optimum gradient consisted in a linear increase of MeOH from 10 to 40% in the first 2 min, increasing to 60% in the next 2 min and finally increasing to 95% in the last 2 min, to a total time of 6 min. Then, 95% MeOH was hold for 1 min in order to clean the column before returning to initial conditions. Under these conditions, the 11 drugs were separated in less than 6 min with a total run time of 8.5 min.
It was observed that at low MeOH proportions in the reconstitution solution glibenclamide and fluvastatin, the most non-polar analytes, presented a weak signal, probably due to their poor solubility in a highly polar environment. On the other hand, for high concentrations of MeOH, the chromatographic peaks of hydrochlorothiazide and amiloride, early eluting analytes, became broader and lost symmetry because of the difference between the eluotropic strength of the sample solution and the starting conditions of the gradient (Fig. 3). The shape of these peaks was also affected by the injection volume, as an increase in the injection volume resulted in an increase of the amount of MeOH injected with the analytes. The best chromatographic performance, avoiding both symmetry and intensity problems, was achieved using a 55:45 aqueous mobile phase:MeOH (v:v) reconstitution solution and a 2.5-µL injection volume. In Fig. 4, the PDA and fluorescence chromatograms obtained with the optimized conditions can be observed.

Effect of methanol percentage of reconstitution solution on the chromatographic separation: 20% (red), 45% (blue) and 80% (black)

PDA (a) and two-channel fluorescence (b) chromatograms obtained for a 5-mg L−1 standard solution under optimized conditions. Hydrochlorothiazide (1), amiloride (2), furosemide (3), bisoprolol (4), rosiglitazone (5) verapamil (6), acenocoumarol (7), valsartan (8), warfarin (9), glibenclamide (10) and fluvastatin (11)
Validation
Recovery, Calibration Range, Accuracy, Precision, and Selectivity
All the analytes showed an acceptable recovery above 75% except amiloride (Table 3), whose poor recovery may be due to the strong interaction with the stationary phase of the cartridge.
The calibration ranges for each of the analytes are collected in Table 3. Intra- and inter-day accuracy and precision %RE and %RSD values for the calibration standards were below the acceptance limits for both the LLOQ (%RE and %RSD <20%) and the rest of the calibration points (%RE and %RSD <15%). The analyzed blank plasmas presented peak area values lower than 20% of the LLOQ areas at the analytes retention times, and lower than 5% of the IS area at its corresponding retention time. Representative chromatograms obtained from a blank human plasma and a plasma spiked with 0.3 mg L−1 of each analyte, and 1 mg L−1 of the IS are shown in Fig. 5.

PDA (a) and fluorescence (b, c) chromatograms obtained for a blank plasma sample (gray) and a 0.3-mg L−1 spiked plasma sample (black) under optimized conditions and treated as explained in point 2.5. Hydrochlorothiazide (1), amiloride (2), furosemide (3), bisoprolol (4), rosiglitazone (5) verapamil (6), acenocoumarol (7), valsartan (8), warfarin (9), glibenclamide (10) and fluvastatin (11)
Robustness of the Method
From the analysis of effects obtained for a confidence level of 95%, it was evidenced the influence of the pH on the resolution of rosiglitazone and verapamil. A negative effect of pH was observed, meaning that the resolution decreases with the pH increase, due to the low polarity of rosiglitazone at high pH values. As a consequence of this change in polarity, the retention time of rosiglitazone gets closer to verapamil, diminishing the resolution between them.
Moreover, the composition of the organic phase in the reconstitution solution has a significant positive effect on the area of glibenclamide and fluvastatin. The area of these analytes increases with the increase in MeOH proportion until reaching 45%, when the response of both analytes stabilizes. Thus, the proportion of MeOH was kept at 45%, taking into account that an increase over this value would affect the shape of hydrochlorothiazide and amiloride.
Analysis of Real Samples
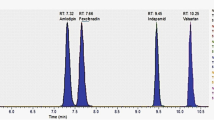
The developed method was applied to plasma samples obtained from patients under cardiovascular treatment with acenocoumarol, furosemide, valsartan or a combination of them. Drug plasma concentration values calculated for clinical samples were obtained by interpolation from the calibration curve. A chromatogram corresponding to each one of the detected analytes is shown in Fig. 6. No interferences between analytes and co-administered drugs were observed which demonstrates the high selectivity of the proposed method.

Chromatograms obtained for a blank plasma sample (gray) and a plasma sample from patients under treatment (black) with: a furosemide (calculated concentration 0.4 ± 0.1 mg L−1), b acenocoumarol (calculated concentration 0.10 ± 0.1 mg L−1) and c valsartan (calculated concentration 1.6 ± 0.2 mg L−1). Furosemide and valsartan were quantified using fluorescence detection and acenocoumarol using PDA detection
Discussion
The use of combined cardiovascular therapy requires the development of reliable analytical methods for the quantification of complex mixtures of drugs in biological matrices. Here, a straightforward method has been developed for the quantification of some of the most prescribed cardiovascular drugs using a UHPLC–PDA–FLD system. Method optimization is an essential step in the pursuance of a suitable quantification method, but usually the time that can be invested in this process is limited. The pharmaceutical industry, as well as the clinical practice, demands methods that can be developed in a fast, easy and simple way. Usually, in many research laboratories, optimization processes are based on previously developed methods or are carried out following OVAT strategies [25, 26]. These can be convenient alternatives, especially if the chromatographic system is coupled to a very selective detector such as a mass spectrometer. However, when the detector is less selective (UV, diode array, etc.) and a complete chromatographic separation is required, those options can be tedious and time-consuming. Furthermore, as interactions among variables are not considered, the obtained results may not be the most advantageous [21]. Experimental design is a very useful alternative to OVAT approaches that takes into consideration the different influential variables and their interactions, and it has proved to be suitable for the optimization of liquid chromatographic separations [15,16,17]. Nevertheless, the application of experimental design to method optimization presents some difficulties that make some researchers reluctant to use it: it requires previous information on the behaviour of the analytes to fix the limits of the domain, specific software and advanced knowledge of statistics, since data interpretation can be misleading, especially if not all the experiments have been carried out properly.
An alternative to these optimization strategies has been applied in this study, following a systematic approach that at the same time takes into account the most important variables in liquid chromatography: the pH of the mobile phase, the nature of the organic modifier and the stationary phase of the chromatographic column.
The application of this procedure for the analysis of the different families of cardiovascular drugs reduces the time required for method development and optimization without requiring complex experimental designs and data processing. The proposed systematic approach proved to be an ideal tool for the optimization of complex chromatographic separation.
This optimization approach can be applied to different drugs and chromatographic techniques. Anyway, the working procedure must be carefully selected in each case. For example, if none of the variable combinations assessed offers a suitable separation, different columns or mobile phases should be taken into consideration.
This systematic approach, halfway between the traditional OVAT optimization and the experimental design, may not offer the total simplicity of the first one nor the immense information of the second one, but has proved to be a very useful alternative for method development. Here, it has been successfully applied to the development of a quantitative method for the analysis of 11 cardiovascular drugs using UHPLC–PDA–FLD, an analytical technique that requires the complete chromatographic separation of the analytes. This method has proved to be adequate for its purpose, fulfilling the accuracy, precision, and selectivity requirements for the validation of bioanalytical techniques. During the validation step, the robustness of the method, a traditionally overlooked parameter, has been thoroughly studied, showing the importance of carefully controlling small changes in the variables that are critical during the optimization step in order to avoid undesired effects on the chromatographic separation. Finally, the method has been applied to samples obtained from patients under cardiovascular treatment demonstrating that it can be used to quantify the analytes of interest in real samples.
References
World Health Organization (2015) Fact sheet: noncommunicable diseases. http://www.who.int/mediacentre/factsheets/fs355/en/. Accessed May 2015
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S (2016) Circulation 133:e38–e360
Lafeber M, Spiering W, Visseren FL, Grobbee DE (2016) Curr Hypertens Rep 18:40
Barrios V, Escobar C (2016) Future Cardiol 12:181–196
Baranowska I, Wilczek A, Baranowski J (2010) Analy Sci: Int J Jpn Soc Anal Chem 26:755–759
Dinç E, Ertekin C, Rouhani G (2015) J Liq Chromatogr Relat Technol 38:970–976
Dobricic V, Vulovic-Tadic M, Jancinc-Stojanovic B, Vladimirov S, Cudina O (2016) Chromatographia 79:571–579
Joshi SJ, Karbhari PA, Bhoir SI, Bindu KS, Das C (2010) J Pharm Biomed Anal 52:362–371
Gonzalez O, Ferreiros N, Blanco ME, Alonso RM (2015) Bioanalysis 7:2399–2417
Baranowska I, Magiera S, Baranowski J (2013) J Chromatogr B 927:54–79
Soltani S, Ramezani AM, Soltani N, Jouyban A (2012) Bioanalysis 4:2805–2821
Lasky T, Greenspan J, Ernst FR, Gonzalez L (2011) Clin Ther 33:2082–2088
Dias E, Hachey B, McNaughton C, Nian H, Yu C, Straka B, Brown NJ, Caprioli RM (2013) J Chromatogr B 937:44–53
Magiera S, Baranowska I, Kusa J (2012) Talanta 89:47–56
Andre C, Jorge F, Castanheira I, Matos A (2013) J Chemom 27:91–98
Zhou Y, Song J-Z, Choi FF-K, Wu H-F, Qiao C-F, Ding L-S, Gesang S-L, Xu H-X (2009) J Chromatogr A 1216:7013–7023
Baranowska I, Magiera S, Baranowski J (2011) J Chromatogr B 879:615–626
Snyder L, Kirkland J, Dolan J (2009) Introduction to modern liquid chromatography. Wiley, New Jersey
European Medicines Agency (2011) EMEA/CHMP/EWP/192217/2009, guideline on bioanalytical method validation
US Department of Health and Human Services, Food and Drug Administration (2001) Guidance for industry: bioanalytical method validation
Esbensen K (2001) Multivariate data analysis in practice. CAMO, Oslo
Dejaegher B, Vander Heyden Y (2007) J Chromatogr A 1158:138–157
Vander Heyden Y, Nijhuis A, Smeyers-Verbeke J, Vandeginste BGM, Massart DL (2001) J Pharm Biomed Anal 24:723–753
Vander Heyden Y, Questier F, Massart DL (1998) J Pharm Biomed Anal 17:153–168
W-d Zhang, W-j Yang, Wang X-j GuY, Wang R (2011) J Chromatogr B 879:3735–3741
Tan KL, Ankathil R, Gan SH (2011) J Chromatogr B 879:3583–3591
Acknowledgements
Authors thank Ministry of Economy and Competitiveness (Project CTQ2013- 46179R), University of Basque Country (GIU 16/04 and UFI 11/23) for financial support and the pharmaceutical companies for the kind supply of studied drugs. M. E. Blanco also thanks the University of the Basque Country (UPV/EHU) for her postdoctoral grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
González Mendia, O., Blanco, M.E., Rico, E. et al. Efficient Method Development and Validation for the Determination of Cardiovascular Drugs in Human Plasma by SPE–UHPLC–PDA–FLD. Chromatographia 80, 605–615 (2017). https://doi.org/10.1007/s10337-017-3274-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-017-3274-6