
Abstract
In spite of lutein and its isomer zeaxanthin being richly available in natural sources, the role of these components on reduction of age-related macular degeneration, cancer, and cardiovascular disorders suggested that an update of the analytical procedure is required to determine the oxidative products and to understand their nutritional significance. In the present study, we have standardized and developed an improved method to obtain characteristic ions of lutein, zeaxanthin, and its major oxidative products in vivo (rats) using LC–MS (APCI)+. In addition, lutein and zeaxanthin isomer were separated on a C30 column with shorter run time with high resolution and calibrated on the basis of picomolar concentration on HPLC (DAD), with the lower detection limit of 0.125 for lutein and 0.128 pmol for zeaxanthin. Characteristic mass spectral ion for lutein is m/z 568.7 [M]+ and 551.5 [M + H–H2O]+ and for zeaxanthin isomer is m/z 568.8 [M]+, 569.8 [M + H]+. Further, optimized conditions produced structurally characteristic fragmented ions under standardized MS (APCI)+ conditions. Total ionic chromatogram together with fine UV–Visible and mass spectra were used to differentiate lutein isomers and its oxidative products, such as 523 [M+ + H+–3CH3], 479 [M+ + H+–6CH3], 551 [M+ + H+–H2O], 276.43 [M+–C22H19O], di-epoxides and 3′-oxolutein. The APCI mass spectral characteristics of major oxidative products of lutein in adult rat tissues are reported here for the first time, to our knowledge. These findings could provide new insights into lutein bioavailability and bioconversions with respect to health benefits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lutein (L) and its stereoisomer zeaxanthin (Z) are members of the xanthophylls family of carotenoids. Among foodstuffs, dark green leafy vegetables, fruits, and egg yolks are rich sources of these pigments [1], and are powerful antioxidants that play an important role in ocular health, prevention of cardiovascular disease, and certain types of cancers [2–4]. The chemical structure of L and its isomers are shown in Fig. 1. Epidemiological studies have suggested that complete characterization of carotenoids and their metabolites in blood and tissues may help in understanding their association with various health benefits. Several analytical methods are available for analysis of carotenoids by HPLC with UV–Visible detection [5]. Further, photodiode array detection assisted by spectral data is the most applied analytical procedure [6]. Many analytical methods were attempted to determine L and its isomers/oxidative products/fragmented ions in food and biological samples by HPLC, LC/MS, and NMR techniques [7–10]. LC–MS analysis of carotenoids was done either with APCI or ESI methods. These techniques may provide comparable sensitivity at a low picomole range and produce abundant molecular ions. While ESI form primarily molecular ions showing poor fragmentation, in contrast APCI produces molecular ions and protonated molecules with higher linearity of detection over a range of lower carotenoid concentration [5]. Apart from determination of L level in natural and biological samples, identification of its oxidative products/fragmented ions at picomolar concentration is a major task in carotenoids biochemistry. Although, these carotenoids are accumulated in the tissues, relatively few studies were made on the formation of oxidative products/fragmented ions in eye samples of human and mice [9, 10]. Therefore, to understand carotenoid metabolism in a methodical manner, an attempt was made further to study the L biotransformation at different tissues using rodents (rats) as model systems. Against this background, using a recently developed HPLC (DAD)–MS + standardized conditions of our research laboratory, we illustrate the detection of isomers/oxidative products/fragmented ions of dietary L in rat plasma and tissues.

Chemical structure of lutein and its major isomer
Materials and Methods
Chemicals
Standard L (99 %), dl-α-tocopherol, and phosphatidylcholine (99 %), were purchased from Sigma-Aldrich (St Louis, MO, USA). Z (99 %) was a kind gift from Dr. V. Baskaran (CFTRI, Mysore, India). HPLC grade acetonitrile, hexane, methanol, dichloromethane, and ammonium acetate were purchased from Sisco Research Laboratories (Mumbai, India). Other chemicals and solvents of analytical grade were purchased from E-Merck Co, Ltd. (Mumbai, India). Animal feed (Amrut feeds, Sangli, India), Casein (Nimesh Corp., Mumbai, India), vitamins and minerals (Hi-Media Laboratories, Mumbai, India) were of high purity and food grade. Olive oil (Leonardo, Nicola Pantaleo Spa, Fasano, Italy) was obtained from a local super market.
Preparation of L for Feeding Trails
A portion (50 mL) of crude acetone extract of green leafy vegetables (GLVs) Trigonella foenum-graecum was evaporated to dryness under N2 gas, the residues redissolved in a known volume (15 mL) of methanolic KOH (10 %), and the extract was saponified overnight under dark conditions at room temperature (25 ± 3 °C). L and Z were extracted with 50 mL of hexane three times or until sample turned colorless. Pooled hexane extracts were evaporated and redissolved in 5 mL of hexane and subjected to column purification on activated neutral alumina [11]. Further, column was washed with hexane until fractions turned colorless and samples were continuously monitored by spectrophotometric measurements. Followed by L/Z a rich fraction was eluted with methanol/dichloromethane (1:1, by vol) and its purity was confirmed by λ max, UV–Visible, and mass spectrum and compared with reference standards. The purified L was further quantified by HPLC (DAD) and used for feeding trails.
Animal Experiments
Animal experiments were performed after due clearance from the institutional animal ethical committee. Two groups (n = 6/group) of male albino rats weighing 250 ± 5 g were housed individually in steel cages at room temperature (28 ± 2 °C) with a 12 h light/dark cycles. Rats were acclimatized for 7 days on a fresh pellet diet and water (Amrut Feeds, Sangli, India), and an isocaloric diet that contained 10 % fat from olive oil was prepared according to AIN-93 (AIN-93). During the 4-week feeding trial, rats were given ad libitum access to freshly prepared diet and had free access to tap water. The purified L was dispersed in olive oil and fed to the animals once in 2 days for 15 doses (0.5 mL/rat). The amount of L fed was 0.445 mg/day/kg body wt and was comparable to the human daily intake [12]. The daily food intake and weekly gain in body weight were monitored. At the termination of the experiment, rats were anesthetized with diethyl ether and sacrificed, and their blood, liver, intestine, and eyes collected. All the samples were processed immediately for L and its isomers/oxidation products/fragmented ions or stored at −80 °C until analysis.
Extraction of L Isomers/Oxidized Products/Fragmented Ions from Biological Samples
L and its isomers/oxidized products/fragmented ions were extracted from plasma, liver, eye, and intestine samples according to the previous procedure with slight modifications [13]. Briefly, to the plasma (0.8 mL), 3 mL of dichloromethane/methanol (2:1, by vol) containing α-tocopherol (2 mM) was added and mixed using a vortex mixer. To the mixture, hexane (1.5 mL) was added, mixed well, centrifuged at 1,000×g for 5 min at 4 °C, and the resulting upper hexane/dichloromethane phase was collected. The extraction procedure was repeated three times with 1 mL of dichloromethane and 1.5 mL of hexane. The extracts were pooled, evaporated to dryness using nitrogen gas, redissolved in 100 μL of mobile phase [acetonitrile/methanol/dichloromethane (60:20:20, v/v/v)], and analyzed by HPLC and LC–MS (APCI)+. Similarly, liver, eye, and intestine samples were homogenized (10 %) (Potter-Elvehjem Homogenizer, Remi Instruments Ltd., Mumbai, India) separately with ice-cold isotonic saline. The 10 % homogenate (0.8 mL) was used for extraction of L and its isomers/oxidative products/metabolites according to the procedure described in the plasma. In case of the liver sample, extract was saponified separately with 2 mL of 10 M methanolic KOH at 60 °C for 45 min in the dark. The reaction mixture was vortexed every 15 min during saponification with an addition of 2 mL of ice-cold deionized water before extraction mentioned elsewhere and analyzed.
HPLC Analysis of L and Its Oxidation Products
Concentration of purified L and Z levels in biological samples (plasma, liver, intestine and eye) was determined by HPLC (DAD, Shimadzu, Japan) [13]. HPLC system is consisting of an LC-10AD pump with SPD-10A UV–Visible absorbance detector (Shimadzu, Kyoto, Japan) and personal computer equipped with Ezchrome chromatography data system software (Scientific Inc., Pleasanton, CA). L was quantified on column and further separation of isomers and oxidation products was achieved through C30 column (5 μm; 250 × 4.6 mm; Princeton, Cranbury, USA), separately. Acetonitrile/methanol/dichloromethane (60:20:20, by vol) containing 0.1 % ammonium acetate was used as a mobile phase. An isocratic analysis was performed at a flow rate of 1 mL min−1 at 450 nm. The components were quantified from their peak area using respective standard curves. The peak identities and λ max of carotenoids were confirmed by their retention time and characteristic spectra of standard chromatograms recorded under a similar condition.
Optimization of LC–MS Condition for L, Z and Its Oxidation Products
LC–MS was performed on a Waters 2996 modular HPLC system with autosampler, gradient pump, thermoregulator, and diode array detector (DAD) coupled to a Q-TOF Ultima (UK) mass spectrometer. The atmospheric pressure chemical ionization (APCI)+ source were heated at 130 °C, and probe was kept at 500 °C. The corona voltage was optimized to 5 kV, HV lens to 0.5 kV, and cone voltage to 30 V. Nitrogen was used as a sheath and the flow rate of drying gas was 100 and 300 L/h, respectively. Mass spectra of L/Z and its oxidative products were acquired with a m/z 0–1,200 scan range, and the UV absorption was recorded at 450 nm using a DAD. The MS identities of L and Z in samples were confirmed using respective reference standards. Due to unavailability of reference standards for L/Z oxidative products and constraint in the animal sample size, the possible structure and configuration of L/Z oxidation products were predicted and characterized using Chemsketch 8.0 software (ACD Labs, USA). The delay time from PDA to the mass spectrometer was observed to be 0.10 min. Data were processed with Mass Lynx 3.2 software. For LC–MS analysis of L/Z and its oxidative products, the mobile phase conditions were mentioned in HPLC analysis section without 0.1 % ammonium acetate. All the isolation, extraction, and analysis of samples were done under dim light to prevent photooxidation.
LC–MS Calibration
The software provides peak integration of the ion abundance for each of the programmed mass ranges. Automated analysis was followed by manual confirmation of fragmentation patterns. TDC (time-to-digital converter) conditions were maintained at centroid thresh hold 1.0, resolution 4,000, and mass window ±1.0. TDC start at 700 (m/V) and TDC stop at 200 (m/V) with threshold 0.00, Pirani Pressure 1.59e0 (mBar) and TOF penning pressure 3.52e−7 (mbar), TDC features used 4 GHz, flight tube were recorded as Ltelf = 1,808.91 and Velf = 9,100. During the analysis of samples the following instrumental conditions were adopted: APCI polarity settings as corona set values 9,601.63; cone voltage, 100–153 fluctuations; RF1 lens energy, 40.0; aperture, 10.0; RF2 lens energy, 2; source temperature between 130/125 and 300/300 °C; cone gas flow 28 (L/h), desolvation gas flow ranges between 300 and 312 (L/h), collision energy, 10.09 eV; and ion energy 1.8e.
Validation of L and Z Analysis
A stock solution of 1 mg/mL of standard L was prepared in methanol. Further dilutions were made to attain the concentration ranging from 0.4 to 3.2 ng with the injection volume of 20 μL to obtain the calibration curve. Standard carotenoids stock and working solutions were analyzed immediately or stored at −80 °C. Each concentration was injected in triplicate (n = 3) to check the reproducibility. The method was validated by plotting the linearity, precision, and intermediate precision as the percent relative standard deviation (RSD %), limit of detection (LOD) with signal-to-noise ratio 3:1, and limit of quantification (LOQ) with a signal-to-noise ratio 10:1. Quantification of L and Z from working standard solutions with the mentioned concentrations was used to study the linear dynamic range (R 2) and ensure the performance of the LC–MS analysis.
Results
Determination and Calibration of L and Z Analysis by HPLC (DAD)
HPLC (DAD) analyses showed there is a significant difference between L and Z quantification under standardized conditions. Further, analysis of standard carotenoids on column was 1 pmol equal to mean of 8,895 ± 657 and Z was 9,296 ± 720 HPLC area, respectively (Fig. 2). The limit of detection and limit of quantification of L was 0.125 and 0.801 pmol and Z as 0.128 and 0.758 pmol, respectively, under standardized conditions. The L standard curve has shown least-squares linear regression analysis of the data, providing an excellent coefficient of determination value (r 2 = 0.992) with slope ± Ss 3.31e + 007, intercept ± Si −3.28e + 003, and % RSD was 2.1 for LOD. Similarly, Z has shown least-squares linear regression (r 2 = 0.991) providing an excellent coefficient of determination with slope ± Ss 3.29e + 005, intercept ± Si −3.23e + 004, and % RSD was 2.0 for LOD. The purity of fractionated lutein isolated from GLVs was confirmed by their characteristics UV–Visible spectra, retention time, and HPLC (DAD)–MS analysis before feeding trails [11].

HPLC (DAD) profile with characteristic UV spectra (a), APCI+ mass spectra (b, c) for lutein and zeaxanthin
MS (APCI)+ Optimization for the Detection of L Fragmented Ions
LC–MS was performed under calibrated conditions coupled to a Q-TOF Ultima mass spectrometer. The conditions adopted were found to be suitable for the detection of abundant L fragmented ions as shown in Table 1. The positive molecular ions were initially acquired full-scan mode with 0.2-step size and 2-ms dwell time and SIM was performed with a dwell time of 200 ms. In goal of L oxidative products characterization, the conditions of analysis are optimized with APCI+ source was heated at 130 °C, and probe was kept at 500 °C. In addition, vaporizer temperature, drying gas temperature, and corona current were all optimized about maximum signal intensity of molecular ions of L oxidative products in plasma and tissue tissues of adult rats (Table 1). Characteristic mass spectral ion for L is 551.5 [M + H–H2O]+ and for Z-isomer m/z 569.8 [M + H]+ % abundance of L fragmented ions was significant at collision energy 10.0 eV. TICs together with fine UV–Visible spectra and mass were used to differentiate possible structural and geometrical isomers and their major oxidative products 523 [M++H+–3CH3], 479 [M+ + H+–6CH3], 551 [M+ + H+–H2O], 276.43 [M+–C22H19O] and L di-epoxides were identified in plasma and liver. Mass spectra of L and Z were acquired with a m/z 0–1,200 scan range, and the delay time from PDA to the mass spectrometer was observed to be 0.10 min.
Characterization of Possible L Oxidation Products/Fragmented Ions in Rat Plasma and Tissues
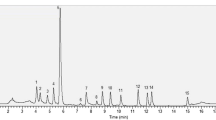
This study developed the efficient extraction and characterization of L oxidation products/isomers in plasma and tissues of adult rats by HPLC–LCMS analysis (Fig. 3). The L level in plasma, liver, intestine, and eye samples, after dietary supplementation, was 50.5 ± 3.0 nmol/L, 161.0 ± 4.0, 230 ± 5, and 140.5 ± 9.3 pmol/g, respectively. The quantitative results are presented as L + Z equivalents, due to some of isomers’ unsatisfactory resolution in plasma and tissue samples. In plasma and liver samples oxidation products were identified and assigned as A, B, C, D, E, F, and G (Fig. 4). Among these, a metabolite eluted at 4.8 min was identified as lutein diepoxide (G), confirmed by UV–Visible spectrum and its characteristic Mass spectra (m/z peak). The chromatographic profile of L oxidative products is well separated with L as a major peak, along with diepoxide and zeaxanthin. The prominent anhydro-lutein molecule was found only in the intestine; with a retention time of 15.3 min; other fragmented ions obtained were designated as B, D, and E (Fig. 5). The characteristic fragmented ions isolated from eye sample were referred as A, H, I, J, and K. The Z peak is more prominent ions (568.9) in L fed eye samples (Fig. 5). L isomers (3-oxolutein, 13-Z lutein, 9-Z lutein, all-E lutein, all-E zeaxanthin, 3-Epilutein and 13-Z zeaxanthin) isolated from rat eye samples were analyzed by HPLC (DAD) and confirmed by their characteristics UV–Visible spectra (λ max) and elution pattern (Fig. 3) [6, 8]. 3′-Oxolutein was prominent in the pooled rat eye samples (6 pairs) (Fig. 5), but it was detected in very low abundance in the pair of eye samples. The retention times for 3′-oxolutein, L, and Z were 6.3, 9.8, and 10.9 min on C-30 column.

HPLC profiles of lutein and its oxidized products/isomers isolated from the rat tissues. *Refer Table 1 for the possible fragmented ions of C and F

APCI+–MS profiles of lutein and its oxidized/metabolized products isolated from rat plasma and liver. Total ionic chromatographic signal at 450 nm recorded online at a retention time of 11.80 and 14.54 min for plasma and liver, respectively. Refer Table 1 for the possible fragmented ions of A/B, C−G. ‡Refer structures mentioned in Fig. 1. *Refer the structures mentioned in plasma sample

APCI+–MS profiles of lutein and its oxidized/metabolized products isolated from rat intestine, eyes (one pair), and pooled rat eyes (five pairs). Total ionic chromatographic signal at 450 nm recorded online at a retention time of 20.26, and 12.01 min for intestine and eyes, respectively. Refer Table 1 for the possible fragmented ions of A/B, D, H−L. *Refer the structures mentioned in plasma sample in Fig. 4. ‡Refer structures mentioned in Fig. 1
Discussion
Carotenoids are extended system of conjugated double bonds and they are unstable in the presence of light, heat, and oxygen. Therefore, the determination of these carotenoids can be challenging and needs sensitive and selective analytical methods [8]. Food and biological samples often contain other compounds that can interfere with carotenoid or mixture of carotenoids with similar structures [14]. In comparison with the standard, an attempt was made to study the possible biotransformation of purified L in adult rats. The available reports have demonstrated that L/Z may play an active role in reducing age-related macular degeneration and certain types of cancer [1–5]. Evidence on the beneficial effects of carotenoids oxidative products has not been illustrated due to limitations of analytical details. The present study reveals optimization of analytical conditions for the isolation, separation, and characterization of possible L/Z oxidation products in adult rat plasma and tissues generated by possible reaction mechanisms as shown in Fig. 6.

Previously, Khachik et al. [7] described the formation of carotenoids oxidative products/metabolites in human milk and serum by HPLC–MS and LC coupled with NMR spectroscopy. Similarly, HPLC and on-line HPLC–MS and –NMR analysis of L and Z isomers in human ocular tissues samples has been well documented [15, 16]. Although these findings are important for analysis of carotenoids oxidative products/metabolites, they do not elaborate on the MS conditions and their optimization. Later, Bhosale et al. [9, 17] have analyzed and identified 3′-oxolutein and 3-methoxyzeaxanthin in human ocular tissues by LC/MS (APCI)+ with following optimized conditions: corona discharge current 5 µA, cone voltage 80 V, and probe temperature 500 °C. Likewise, Ferreria et al. [18] have conditioned the LC/MS (APCI)+ for the characterization and elucidation of enzymatic and oxidative cleavage products of lycopene under the conditions of APCI corona discharge at 2,200 V, vaporizer temperature 300 °C, nebulizer pressure of 50 psi of N2 and set with 350 °C drying gas temperature optimized at a flow rate of 4.0 L/min. Further, lycopene fragmented ions were detected in the mass scan range between 200 to 700 Da in 0.1 Da with an accumulation time at 50 ms. The mass spectra of geometrical isomers found in tissues of rats were identical and differed only in their chromatographic elution pattern and spectral behavior (shifting of λ max) [19]. In the present study APCI+ source temperature was monitored at 130 °C and probe temperature at 500 °C. In addition, corona voltage was optimized to 5 kV, HV lens to 0.5 kV, and cone voltage monitored with lower 30 V for the identification of L oxidative products in rat plasma and tissues (Figs. 2, 3, 4, 5). Standardization of APCI+ probe temperature and related parameters adopted was shown to be very crucial for better ionization of the L and its fragmented ions. Under the adopted analytical conditions, L shows a base peak at m/z 551.5 despite its equal molecular weight with zeaxanthin, due to a shift in the double bond. L possesses allylic hydroxyl group which allows loss of H2O molecule after protonation under the conditions, which is similar to the earlier report [15]. Under the standardized conditions, we have identified the prominent ions of L-epoxides in plasma and hepatic tissue after oral supplementation of L, in contrast to the previous report which stated that xanthophylls epoxides are not detectable in plasma after oral administration of carotenoids in human subjects [20]. The pattern of L oxidative metabolites formed in the liver was similar to plasma; reflection of results in plasma indicated that diepoxides may be formed in the liver itself and mobilized to the blood (Fig. 6). The chromatographic profile of L oxidative products is well separated with L as a major peak, along with diepoxide and zeaxanthin. Only few reports are available on identification and quantification of epoxy-carotenoid in biological samples [21, 22]. Recently, Yonekura et al. [10] identified keto-carotenoid є,є-carotene-3,3′-dione as a major metabolite of dietary L in mice. However, in Z (3R and 3′R) non-allelic nature of the hydroxyl group at C3 and C3′ may undergo isomerization to yield epilutein before oxidation. The only evidence available in support of this metabolic transformation is the mere presence of 3-hydroxy-lutein, epi-lutein, and meso-zeaxanthin in the eye samples. Further, Prasain et al. [8] identified two oxime derivatives of 3-hydroxy-ionone and 3-hydroxy-14-apocarotenal with protonated molecules at m/z 252 and m/z 370, respectively, in the human eye sample using LC/MS/MS. They also demonstrated the formation of Z and its oxidative products by electrospray ionization interface in the positive mode monitored at 4900 V; the orifice potentials were 60 V (Z) and 40 V for its oxidative derivatives. These results indicate that a fragmentation pattern of carotenoids is varied depending on the conditions adopted and its optimization. In continuation, we also revealed the importance of APCI conditions’ optimization for determination of fragmented ions of L oxidative products and % abundance of their respective ions. In addition, protonated molecules [M + H]+ of carotenoids upon collision-induced dissociation produced a number of characteristic L fragmented ions at collision energy 10 eV. The identification of oxidized carotenoids in biological samples remains a major task due to their less concentration and lower stability. These studies have clearly indicated that the oxidation products of L identified in the biological samples are not due to artifacts and storage, etc. The NMR studies of compounds could not be carried out due to the stability and quantitative limitations [15, 23].
Conclusion
Validation of carotenoids analysis could be improved by substantial modifications of LC–MS conditions. The development of calibrated conditions of liquid chromatography coupled with APCI+–MS analysis allows the separation and identification of 12 possible L oxidative products. In addition, we have achieved the limit of detection at below picomolar range with excellent reproducibility. This is the first report to detect L oxidative products in plasma and tissues of adult rats under the described LC–MS conditions. The detailed characterization of L oxidative products by MS/MS and influence of these compounds on the biological significance with respect to chronic and eye-related degenerative disorders deserve further studies.
References
Sommerburg O, Keunen JEE, Bird AC, van Kuijk FJGM (1998) Fruits and vegetables that are sources for lutein and zeaxanthin: the macular pigment in human eyes. Br J Ophthalmol 82:907–910
Landrum JT, Bone RA (2001) Lutein, zeaxanthin, and the macular pigment. Arch Biochem Biophys 385:28–40
Bowman PE, Mobarhan S (1995) Evidence from cancer intervention and biomarker studies and the development of biochemical markers. Am J Clin Nutr 62:1403S–1409S
Krichevsky SB (1999) β-carotene, carotenoids and the prevention of coronary heart disease. J Nutr 129:5–8
Rezanka T, Olsovska J, Sobotka M, Sigler K (2009) The use of APCI–MS with HPLC and other separation techniques for identification of carotenoids and related compounds. Curr Anal Chem 5:1–25
Britton G (1995) Structure and properties of carotenoids in relation to function. FASEB J 9:1551–1558
Khachik F, Spangler CJ, Smith JC Jr, Canfield LM, Steck A, Pfander H (1997) Identification, quantification, and relative concentrations of carotenoids and their metabolites in human milk and serum. Anal Chem 69:1873–1881
Prasain JK, Moore R, Hurst JS, Barnes S, Van Kuijk FJGM (2005) Electrospray tandem mass spectrometric analysis of zeaxanthin and its oxidation products. J Mass Spectrom 40:916–923
Bhosale P, Zhao DY, Bernstein PS (2007) HPLC measurement of ocular carotenoid levels in human donor eyes in the lutein supplementation era. Invest Ophthalmol Vis Sci 48:543–549
Yonekura L, Kobayashi M, Terasaki M, Nagao A (2010) Keto-carotenoids are the major metabolites of dietary lutein and fucoxanthin in mouse tissues. J Nutr 140:1824–1831
Lakshminarayana R, Raju M, Krishnakantha TP, Baskaran V (2005) Determination of major carotenoids in a few Indian leafy vegetables by high-performance liquid chromatography. J Agric Food Chem 53 8:2838–2842
Richer SW, Stilies L, Statkute J, Pulido J (2004) Double-masked, placebo-controlled, randomized trial of lutein and antioxidant supplementation in the intervention of atrophic age-related macular degeneration: the veterans LAST study (Lutein antioxidant supplementation trial). Optometry 75:216–230
Baskaran V, Sugawara T, Nagao A (2003) A phospholipids affect the intestinal absorption of carotenoids in mice. Lipids 38:705–711
Van Breeman RB (1997) Liquid chromatography/mass spectrometry of carotenoids. Pure Appl Chem 69:2061–2066
Dachtler M, Glaser T, Kohler K, Albert K (2001) Combined HPLCMS and HPLC–NMR on-line coupling for the separation and determination of lutein and zeaxanthin stereoisomers in spinach and in retina. Anal Chem 73:667–674
Bone RA, Landrum JT, Hime GW, Cains A, Zamor J (1993) Stereochemistry of the human macular carotenoids. Invest Ophthalmol Vis Sci 34:2033–2040
Bhosale P, Bernstein PS (2005) Quantitative measurement of 3′-oxolutein from human retina by normal-phase high-performance liquid chromatography coupled to atmospheric pressure chemical ionization mass spectrometry. Anal Biochem 345:296–301
Ferreira ALDA, Yeum KJ, Russell RM, Krinsky NI, Tang G (2004) Enzymatic and oxidative metabolites of lycopene. J Nutr Biochem 15:493–502
Emenhiser C, Sander LC, Schwartz SJ (1995) Capability of a polymeric C30 stationary phase to resolve cis–trans carotenoid isomers in reversed-phase liquid chromatography. J Chromatogr A 707:205–216
Barua AB, Olson JA (1998) Reversed-phase gradient high-performance liquid chromatographic procedure for simultaneous analysis of very polar to nonpolar retinoids, carotenoids and tocopherols in animal and plant samples. J Chromatogr B 707:69–79
Bausch J, Licchite H, Oesterhelt G, Kister A (1999) Isolation and identification of a major urinary canthaxanthin metabolite in rats. Int J Vitam Nutr Res 69:268–272
Asai A, Terasaki M, Nagao A (2004) An epoxide furanoid rearrangement of spinach neoxanthin occurs in the gastrointestinal tract of mice and in vitro: formation and cytostatic activity of neochrome stereoisomers. J Nutr 134:2237–2243
Aman R, Biehl J, Carle R, Conrad J, Beifuss U, Schieber A (2005) Application of HPLC coupled with DAD, APCI–MS and NMR to the analysis of lutein and zeaxanthin stereoisomers in thermally processed vegetables. Food Chem 92:753–763
Acknowledgments
Authors acknowledge the Department of Biotechnology, Bangalore University, for their encouragement and support. R. Lakshminarayana acknowledges the University Grant Commission, New Delhi, India, for the grant of Major Research Project (F.No.37-84/2009-SR), UGC-SAP and DST-FIST facilities and support. P. R. Sowmya acknowledges the grant of Women Scientist Fellowship by Department of Science and Technology (New Delhi, India).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sowmya, P.R., Arathi, B.P., Vijay, K. et al. Optimization of LC/MS (APCI)+ Methods for the Determination of Possible Lutein Oxidation Products in Plasma and Tissues of Adult Rats. Chromatographia 77, 1633–1642 (2014). https://doi.org/10.1007/s10337-014-2765-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-014-2765-y