
Abstract
Haemosporidian parasites (genera Plasmodium and Haemoproteus) are common blood parasites of birds transmitted by dipteran insect vectors. We analyzed blood samples from 329 individuals of 43 bird species in eastern Tennessee to better understand the relationship between the local community of birds and their blood parasites, including the distribution of parasites across hosts and the underlying ecological factors and life -history traits that influence parasite prevalence across host species. Using molecular methods, we found 144 individuals of 25 species to be infected with haemosporidian parasites (overall prevalence of 44 %). We distinguished 22 genetic lineages, including 11 in the genus Haemoproteus and 11 in Plasmodium. Fourteen percent of infected individuals harbored more than one parasite lineage. Across species, total prevalence increased with local abundance and decreased with incubation period, but did not vary with nesting or foraging height, average annual survival of host species, migratory or flocking behavior, sexual dimorphism, average species mass, or among sites. The prevalence of Haemoproteus was higher in species that nest 1–5 m above ground than in species that nest below 1 m or above 5 m, and the prevalence of Plasmodium was marginally higher in species with open-cup nests. Infection status did not vary with age, sex, or body condition. Our research reveals substantial variation in prevalence and richness of haemosporidian parasites, some of which is related to specific avian life history traits.
Zusammenfassung
Prävalenz von aviären Hämosporidien in Osttennessee und deren Beziehung zur Biologie der Wirtsvögel
Hämosporidien der Gattungen Plasmodium und Haemoproteus sind bei Vögeln verbreitete Blutparasiten, welche von zweiflügeligen Insekten (Diptera) als Vektoren übertragen werden. Für ein besseres Verständnis der Beziehung zwischen einer lokalen Vogelgemeinschaft und deren Blutparasiten, einschließlich der Verteilung der Parasiten auf die Wirtsorganismen und die zugrunde liegenden ökologischen und biologischen Faktoren, welche die Prävalenz der Parasiten bei den Wirtsarten beeinflussen, analysierten wir Blutproben von 329 Individuen aus 43 Vogelarten in Osttennessee. 144 Individuen aus 25 Arten waren von hämosporidischen Parasiten befallen (Gesamtprävalenz 44 %); mithilfe molekularer Methoden ließen sich 22 genetische Stämme unterscheiden; von diesen gehörten elf zur Gattung Haemoproteus und elf zu Plasmodium. 14 % der infizierten Individuen wiesen mehr als einen Parasitenstamm auf. Bei allen Arten stieg die Gesamtprävalenz mit der lokalen Häufigkeit an und nahm mit der Brutdauer ab, variierte aber nicht in Abhängigkeit von der Nesthöhe oder der Höhe der Futtersuche, der durchschnittlichen jährlichen Überlebensrate der Wirtsart, dem Zug—oder Schwarmverhalten, Geschlechtsdimorphismen, der Durchschnittsmasse der Art oder in Abhängigkeit vom Ort. Die Prävalenz von Haemoproteus war höher bei Arten, die zwischen 1–5 m über dem Erdboden nisteten als bei solchen, welche entweder unter 1 m oder über 5 m nisteten; die Prävalenz von Plasmodium lag bei Arten mit offenen Napfnestern geringfügig höher. Der Infektionsstatus variierte weder mit dem Alter, dem Geschlecht noch mit der Kondition. Unsere Untersuchung belegt eine beträchtliche Variation bezüglich der Prävalenz und Vielfalt hämosporidischer Parasiten, die teilweise von der spezifischen Lebensweise der Vogelarten abhängt.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parasites influence individuals, populations, and communities of hosts in several ways. They can affect host behavior (Thomas et al. 2002; Møller 2008), dispersal (Brown and Brown 1992) and fitness (Fitze et al. 2004; Marzal et al. 2005; Knowles et al. 2010). Parasites can also influence the structure of their host communities, often by their indirect influence on trophic interactions (Lafferty et al. 2006, 2008), competitive interactions, and energy flow throughout a community (Hudson et al. 2006), and by constituting a large part of its biomass (Kuris et al. 2008). Parasites may also drive sexual selection in their hosts (Hamilton and Zuk 1982).
Haemosporidian blood parasites (informally known as avian malaria parasites; Pérez-Tris et al. 2005) in the genera Plasmodium and Haemoproteus (Haemosporida: Plasmodiidae and Haemoproteidae, respectively) are common, widespread parasites of birds (Valkiūnas 2005). Their avian hosts are most affected during the acute phase of the infection (Ferrell et al. 2007). Individuals that survive this phase enter the chronic phase, when parasitemia (i.e., parasite intensity) levels are lower. Such individuals, however, are still infected and thus able to transmit parasites through vectors (Valkiūnas 2005). The impact of haemosporidian infection can be lineage-specific (Lachish et al. 2011) and can have dramatic consequences for evolutionary naïve host communities, such as native Hawaiian birds (van Riper et al. 1986).
Haemosporidian parasite prevalence varies widely across host species and can vary with host density (Ricklefs et al. 2005; Isaksson et al. 2013) and ecological and life history traits (Fecchio et al. 2013; Svensson-Coelho et al. 2013; Lutz et al. 2015). Plasmodium and Haemoproteus parasites reproduce sexually in dipteran vectors—culicid mosquitoes and ceratopogonid midges, respectively (Valkiūnas 2005)—and use birds as intermediate hosts in which they undergo asexual reproduction. Plasmodium vectors are primarily distributed near the ground, while Haemoproteus vectors are more abundant near the canopy (Cerný et al. 2011). This distinct spatial distribution of vectors suggests that parasites transmitted by these vectors may vary across host species that nest or forage at different heights. Fecchio et al. (2011) found that the influence of nest type on parasite prevalence varied by vector: Plasmodium prevalence was higher in host species with closed-cup nests, while the reverse was true for Haemoproteus. Results related to Plasmodium could be due to an accumulation of carbon dioxide, an olfactory cue used by vectors (Gibson and Torr 1999) to locate hosts in closed spaces (Fecchio et al. 2011). Conversely, Ribeiro et al. (2005) suggested that vectors might more likely come into contact with species that nest in open-cup nests due to increased exposure, implying that open-cup nesters may be more susceptible to haemosporidian parasites than cavity nesters.
In addition to species-level ecological factors leading to differences in infection status, evolutionary factors may also be important. For example, sexual dimorphism may be related to parasite prevalence as a result of sexual selection favoring costly male phenotypic traits as indicators of parasite resistance (Hamilton and Zuk 1982). In support of this idea, some analyses have found a positive association between parasite prevalence and plumage brightness (Read 1987; Doucet and Montgomerie 2003; Scheuerlein and Ricklefs 2004), although others have failed to find such a relationship (Borgia and Collis 1989; Ricklefs et al. 2005; Bensch et al. 2007).
Individual-level host traits, such as age, sex, and body condition, may also influence infection status in varying ways. For example, one hypothesis holds that parasite prevalence might be higher in older birds as a result of accumulated exposure to parasites (Wilson et al. 2002; Ricklefs et al. 2005). The relationship between infection status and age might be more complicated if specialist and generalist parasites infected host individuals primarily in older and younger age classes, respectively (Medeiros et al. 2014). Similarly, infection status could be influenced by sex, with males experiencing higher susceptibility to parasites, possibly as a result of the energetic trade-off between immunocompetence and reproductive effort (Norris et al. 1994; Richner et al. 1995; Zuk and McKean 1996). Norte et al. (2009) suggested that females experience more costly energetic constraints on immune function during the incubation period, while males experience depressed immune function during the brood-rearing period. Such partitioning of reproductive investment could influence the appearance of malaria infections in the circulating blood. Infection status may also be influenced by body condition, with infection or failure to control infection more likely among birds in poorer condition (Dawson and Bortolotti 2000; Norte et al. 2009).
Haemosporidian parasites provide an excellent model system for studying host–parasite interactions, because the parasite lineages are dispersed on a global scale and their effects can be easily studied in both observational and experimental settings (Marzal et al. 2005; Valkiūnas 2005; Fallon et al. 2006). Studies of the distribution of haemosporidian blood parasites across host species and the relationship of infection to host life histories in localities in eastern North America have been sparse (Garvin et al. 1993; Schrader et al. 2003; Ricklefs et al. 2005; Astudillo et al. 2013). This study seeks to increase our understanding of factors that influence patterns of haemosporidian infection in local avian communities. Here, we quantify haemosporidian prevalence across individuals and species, and hypothesize that (1) infection status is related to individual-level traits (e.g., age, sex, and body condition) that may indicate variation in host immune defense mechanisms, and that (2) haemosporidian prevalence is related to species-specific life history and ecological traits of hosts (e.g., nesting and foraging heights, nest type, abundance, flocking behavior, migratory status, and incubation period), which are in turn related to differential dipteran vector exposure and to transmission of infection.
Methods
Study sites
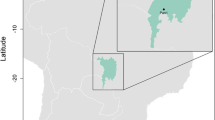
From May 22 to June 1, 2013, we sampled birds during the breeding season at four sites in eastern Tennessee: Seven Islands Wildlife Refuge (35°57′12.73″N, 84°41′33.99″W and 35°57′12.73″N, 84°41′33.99″W), Kyker Bottoms Refuge (35°36′07.27″N, 84°07′17.26″W; 35°36′07.27″N, 84°07′17.26″W; and 35°36′15.95″N, 84°07′27.57″W), Ijams Nature Center (35°57′14.73″N, 84°51′47.90″W), and Melton Hill Park (35°57′06.45″N, 84°14′07.59″W and 35°57′01.27″N, 84°14′07.25″W) (Fig. 1). These sites are characterized as eastern deciduous forest, and contain a mixture of early-successional and mid- to late-successional stands, with average canopy height of up to 20 m. All sites were located within 500 m of a major body of water, and elevations ranged from 240 to 310 m asl.

Map of field sites (inset). Field sites are denoted by black stars, with major surrounding cities denoted by black dots. Thin lines denote major rivers and streams. This figure was created using ArcGIS version 10.2 software
Field sampling of birds
At each location, we set 10–17 mist nets in locations with high bird activity, often along forest edges, footpaths, or off-road trails. Mist nets were 38-mm gauge, 2.6 m tall, and 6, 9, or 12 m long. We did not bait nets with food. Because parasite infection prevalence can vary by habitat (Loiseau et al. 2010), we set nets in or adjacent to early-successional habitats. We sampled birds between 0540 and 1230, checking nets every 20–30 min. A U.S. Geological Survey band was placed on the left leg of each captured individual. We identified individuals to species, aged and sexed them according to Pyle (1997), and measured mass and wing length. Captured individuals were released at the site after processing. Banding occurred under federal permit #23734 and state permit #3666. Capturing and processing protocols were approved by Rhodes College Institutional Animal Care and Use Committee (IACUC; #111 and 114).
Blood sampling
We obtained ca. 30 μl of blood from the sub-brachial vein (never exceeding 1 % of the individual’s body weight), which was drawn into capillary tubes; 5–10 μl of blood was immediately put into 300 μl of lysis buffer (Longmire et al. 1997). Blood samples were stored at room temperature until DNA extraction in the lab.
Molecular analyses
DNA extraction
We added 5 µl of Proteinase K (IBI Scientific, Peosta, IA, USA) to each blood sample in lysis buffer, and incubated at 60 °C in a water bath for 4–8 h. DNA was then extracted using a standard ammonium acetate-isopropanol extraction (Svensson and Ricklefs 2009), and we checked each extraction for the presence of DNA before screening for infections.
Screening for infections
We screened all DNA samples for infection using a polymerase chain reaction (PCR) protocol designed to amplify a small fragment of the haemosporidian 16S rRNA gene (Fallon et al. 2003). We screened from 4 to 28 samples in each reaction. We used one positive (a known positive malaria infection based on microscopy) and one negative (ultrapure water) control in each reaction, and PCR products were visualized with a 1 % agarose gel stained with ethidium bromide and run for 20 min at 94 V to determine positive infections.
Cytochrome b amplification
For samples that screened positive for the 16S rRNA gene fragment of the parasites, we ran a nested PCR reaction to amplify a ~550-bp fragment of the haemosporidian cyt b gene. The initial outer reaction amplified a ~750-bp fragment of the haemosporidian cyt b gene using primers 3932F (Olival et al. 2007) and DW4R (Perkins and Schall 2002) in the following PCR regime: 94 °C (4 min), 35 cycles of 94 °C (20 s), 49 °C (10 s), 68 °C (45 s), and finally, 68 °C (3 min). In each reaction we used 0.5 μl of DNA, 6.25 μl water, 1 μl MgCl2 free 10X buffer, 0.8 μl dNTPs (2.5 mM), 0.8 μl MgCl2 (25 mM), 0.2 μl 3932F (10 μM), 0.2 μl DW4R (10 μM), 0.2 μl BSA (1X), and 0.05 μl Takara Taq (5U/μl TaKaRa Bio, Shiga, Japan). For the inner reaction, we used primers 413F and 926R (Ricklefs et al. 2005) in the following PCR regime: 94 °C (1 min), 28 cycles of 94 °C (20 s), 52 °C (10 s), 68 °C (50 s), and finally, 68 °C (7 min). In each inner reaction we used 13 μl water, 2 μl MgCl2 free 10X buffer, 1.6 μl dNTP, 1.6 μl MgCl2, 0.4 μl 413F (10 µM), 0.4 μl 926R (10 µM), 0.4 μl BSA (1X), and 0.1 μl Takara Taq, along with 0.5 μl of PCR product from the outer reaction. This protocol is similar to that described in Fecchio et al. (2013). We used one positive and one negative control in each outer reaction and one positive and a negative control after every fourth sample in the inner reaction, including a negative control from the outer reaction.
Sequencing
We purified the PCR products of all positive inner cyt b reactions using the following ExoSAP protocol: 2.6 μl of ultrapure water, 0.2 μl of Antarctic phosphatase (New England Biolabs, M0289L), and 0.2 μl Exonuclease (New England Biolabs, M0293L) to each sample, and incubated at 37 °C for 30 min and at 60 °C for 15 min. Purified PCR samples were sequenced on an Applied Biosystems 3130xl sequencer (Thermo Fisher Scientific, Waltham, MA, USA) at the University of Tennessee Health Sciences Center. All positive infections were sequenced in forward and reverse directions, and contigs were assembled in ChromasPro (Technelysium, Version 1.7.5).
Identifying lineages
We aligned sequences in Mega Version 5.2 (Tamura et al. 2011). Ambiguous areas in the sequences were examined in the chromatograms using ChromasPro. We assumed that double peaks in chromatograms indicated mixed infections in an individual, and we assigned lineage names to the parasites of individuals with mixed infections when possible (Online Resource 1). We phased parasite haplotypes from a mixed infection by assigning ambiguity codes to base pairs that presented multiple peaks in the sequence chromatogram, and then compared the ambiguous sequence to a dataset of known parasite lineages. This process is often employed and rarely explained in the avian haemosporidian literature, and so we explain it in detail in Online Resource 1. Individuals for which we could not assign a distinct lineage were included in analyses of total infection prevalence, and individuals for which we could determine the parasite genus were additionally included in separate analyses of Plasmodium and Haemoproteus. We assigned lineage names to groups of haplotypes with less than 1 % sequence divergence and who had similar host distributions. Unique lineages were compared to lineages deposited in GenBank (available from the National Center for Biotechnology Information at www.ncbi.nlm.nih.gov) and with avian infections from sites in North America (Ricklefs et al. 2014). When a unique lineage had a 100 % match on GenBank or in our database, we renamed the lineage according to the deposited name. We found a single undescribed lineage (TN24), the sequence for which is available, along with all other lineages, on GenBank (Online Resource 2, Table 1).
Statistical analysis
We used generalized linear mixed models (GLMMs) to assess whether individual infection was influenced by individual-level traits such as age, sex, and body condition. Body condition (or size-corrected body mass) is measured as the residuals of a species-specific log(mass) by log(wing length) regression, which is often used as a proxy for overall condition (Schulte-Hostedde et al. 2005). GLMMs were also used to assess whether infection prevalence in each host species was influenced by species-specific traits, including abundance, mean species body mass (g), nest type (open- vs. closed-cup), species average nest height (<1, 1–5, >5 m), species average foraging height (<1, 1–5, >5 m), migratory status, flocking behavior, incubation period, and degree of sexual dimorphism. We were unable to obtain estimates of all predictor variables for all individuals or all species, so degrees of freedom change with the variable examined. In addition, Brown-headed Cowbird (Molothrus ater), a brood parasite, was excluded from analyses of nesting parameters (e.g., nest type, nest height, and incubation period).
We derived abundance estimates for each species from contour abundance maps generated from BBS (Breeding Bird Survey) data and downloaded as shapefiles (available from Patuxent Wildlife Research Center at http://www.mbr-pwrc.usgs.gov/bbs/geographic_information/GIS_shapefiles_2010.html). Sauer et al. (2011) detail how these maps were produced. Briefly, they averaged point counts on each BBS route from the years 2006–2010 and used a distance-weighted average of counts (Isaaks and Srivastava 1989) to estimate the abundance of each species across its breeding range within the continental United States. We imported abundance maps to R v3.0.2 (R Core Team 2013), using the ‘rgdal’ package (Bivand et al. 2013), converted species polygons to rasters using the ‘raster’ package (Hijmans et al. 2014), and then extracted species-specific abundance estimates for our site (see Online Resource 3). Abundance estimates represent the predicted number of individuals of a given species observed in ~2.5 h of surveying (Sauer et al. 2011).
We obtained estimates of annual survival for each species from the MAPS (Monitoring Avian Productivity and Survivorship) Avian Demographics Query Interface (Michel et al. 2011) for the Southeast region. These estimates measure the overall patterns of average annual survival rates of birds in North America (Desante et al. 1995). We obtained estimates of average species body masses from the CRC Handbook of Avian Body Masses (Dunning 2008). When male and female masses were reported separately, we recorded the mean. Each species was categorized by degree of sexual dimorphism: none; intermediate, with phenotypic differences limited to the face and head; and high, with phenotypic differences occurring beyond the face and head. We obtained all other species-level data (nest type, nest height, foraging height, migratory and flocking behavior, and incubation period) from The Birds of North America Online (Rodewald 2015). Table 1 contains all species-level data used in our analyses.
For individual-level traits (e.g., age, sex, and body condition) and site comparisons, we tested patterns of infection for Plasmodium and Haemoproteus both separately and combined, and we examined each predictor separately, because the number of species sampled did not permit a single analysis with all predictor variables examined simultaneously. We used the “GLIMMIX” procedure in SAS 9.3 (SAS Institute Inc. 2011) to run a mixed effect model with bird taxonomy (species nested in genus and genus nested in family) as a random effect. For species-level traits (e.g., abundance, foraging height, and nest type), we examined only species with four or more individuals sampled and used the “GLIMMIX” procedure to examine the effects of those species-level traits on disease prevalence (proportion of individuals within a species that exhibit infection). Because no genus contained more than one species with four or more individuals, we included species nested in family as a random effect.
Results
Prevalence variation
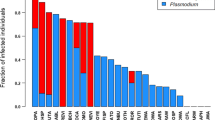
We screened blood samples from 329 individuals of 43 species for avian malaria. Of these, 144 birds (44 %) of 25 species (58 %) were infected. We detected Plasmodium in 95 individuals (29 %) of 18 species (42 %) and Haemoproteus in 55 individuals (17 %) of 18 species (42 %). Plasmodium infections were more frequent than Haemoproteus infections (binomial test, p < 0.01). Among infected individuals, 20 birds (14 %) from ten species exhibited mixed infections. Total prevalence (infection by either Plasmodium or Haemoproteus, or both) varied significantly among species (χ2 = 97.8; df = 19; p < 0.001), ranging from 0 to 100 % (Table 2). Prevalence also varied across species when only well-sampled species with ten or more individuals were examined (χ2 = 69.8; df = 7; p < 0.001), and prevalence ranged from 5.3 % in American Goldfinch (Spinus tristis) to 88.9 % in Northern Cardinal (Cardinalis cardinalis). All species with nine or more sampled individuals showed at least one infection; the most commonly sampled species with no infections was the Tree Swallow (Tachycineta bicolor; 8 individuals).
Across species, there was no relationship between the prevalence of Plasmodium and the prevalence of Haemoproteus (Spearman’s rho = −0.29; p = 0.49). After accounting for across-site differences in species sampled by including taxonomic affiliation as a random effect (with species nested in genus and genus in family), and omitting one site (Ijams Nature Center) due to low sample size, we found no significant heterogeneity in total prevalence (F 2,277 = 1.13; p = 0.33) or in the prevalence of Plasmodium (F 2,277 = 0.21; p = 0.81) or Haemoproteus (F 2,277 = 0.97; p = 0.38) among the three sites included in the analysis.
Likelihood of infection and individual-level traits
Infection status did not vary with age. Hatch Year (HY), After Hatch Year (AHY), Second Year (SY), and After Second Year (ASY) birds did not differ significantly in infection status (F 3,278 = 0.15; p = 0.93), and a comparison between SY and ASY individuals was also non-significant (F 1,98 = 0.01; p = 0.91). Infection status also did not vary with sex (F 1,257 = 0.67; p = 0.41) or body condition (F 2, 277 = 1.13; p = 0.33). With the exception of SY individuals having marginally higher Haemoproteus infection status than ASY individuals (F 1,98 = 4.12; p = 0.045), the infection status for Plasmodium and for Haemoproteus separately also did not vary with age, sex, or body condition (p > 0.15 for all tests).
Influence of species-level traits
Total prevalence increased with log-transformed BBS estimates of species abundance (β = 1.77; F 1,268 = 6.68; p = 0.010; Table 3). Prevalence of Plasmodium (β = 0.91; F 1,268 = 1.78; p = 0.18) and of Haemoproteus (β = 1.83; F 1,268 = 3.31; p = 0.07) showed positive trends but were not significant (Table 3).
Total prevalence (F 1, 266 = 4.33; p = 0.039; Table 3) and prevalence of Plasmodium were higher in open-cup than in closed nests (F 1,266 = 4.62; p = 0.033; Table 3). Haemoproteus prevalence did not differ with nest type (F 1,266 = 0.00; p = 0.94; Table 3).
Total prevalence (F 2,271 = 1.50; p = 0.22) and prevalence of Plasmodium (F 2,271 = 1.92; p = 0.15) did not vary with species average nest height (Table 3). Species that nest at 1–5 m had a higher prevalence of Haemoproteus than species nesting below 1 m or above 5 m (F 2,271 = 5.24; p = 0.006; Fig. 2; Table 3).

Haemoproteus prevalence and nesting height (inset). Species that nest at 1–5 m had a higher prevalence of Haemoproteus than species nesting below 1 m or above 5 m (F 2,271 = 5.24; p = 0.006; Table 3)
Neither total prevalence, prevalence of Plasmodium, nor prevalence of Haemoproteus were related to foraging height (p > 0.20 for all tests), average annual survival (p > 0.60 for all tests), or degree of sexual dimorphism (p > 0.15 for all tests; Table 3).
Prevalence of Haemoproteus increased with average species body mass (β = 3.41; F 1,271 = 7.53; p = 0.007; Table 3), but this pattern did not hold for Plasmodium (β = −0.93; F 1,271 = 0.73; p = 0.39; Table 3) or total prevalence of both genera of parasites together (β = 0.71; F 1,271 = 0.40; p = 0.53; Table 3).
Haemoproteus prevalence was lower in neotropical migrants than in residents or short-distance migrants (F 2,271 = 5.04; p = 0.007; Table 3), but migratory status was not related to either total prevalence (F 2,271 = 0.10; p = 0.91; Table 3) or to prevalence of Plasmodium (F 2,271 = 0.74; p = 0.48; Table 3).
Total prevalence, prevalence of Plasmodium, and prevalence of Haemoproteus were not related to social behavior and did not differ between solitary species and flocking species (species that live in small groups or in small or large flocks) (p > 0.25 for all tests; Table 3).
Total prevalence decreased with incubation period (β = −0.57; F 1,266 = 4.24; p = 0.040; Table 3). Prevalence of Plasmodium (β = −0.46; F 1,266 = 3.50; p = 0.062; Table 3) and prevalence of Haemoproteus (β = −0.20; F 1,266 = 0.40; p = 0.53; Table 3) showed negative but non-significant trends with incubation period.
Our analyses included an introduced species, House Sparrow (Passer domesticus). When this species is excluded, most results remain unchanged. However, total prevalence and prevalence of Plasmodium no longer differ with nest type, probably because of lower sample size among closed-cup nesters and reduced statistical power. Additionally, the negative but non-significant relationship between Plasmodium prevalence and incubation period becomes significant when House Sparrows are removed from the analysis. All other species-level analyses remain qualitatively unchanged.
Host specificity
We recovered 22 cyt b lineages from 144 birds: 11 lineages were Plasmodium and 11 were Haemoproteus. Of the 22 lineages, 16 (73 %) were obtained from at least two individuals. Of the 16 lineages that were obtained at least twice, 13 (81 %) were found in more than one host species (median = 2; mean = 3.5; range 1–12). Each of the nine lineages obtained from only one species was found in only one (6 lineages) or two (3 lineages) individuals. Host species with nine or more sampled individuals harbored between one and seven parasite lineages (Table 2). Twenty individuals from ten species showed mixed infections; from these birds, we found 44 infections and were able to identify the lineage for 37. These 37 infections represented 11 lineages (Table 3), four of which were not recovered from any bird with a single infection. One lineage (CHI35PL) was obtained only from two birds with mixed infections. Another lineage (OZ14) was recovered from five birds with mixed infections (25 % of the total number of birds with mixed infections). In other localities in eastern North America, OZ14 has occurred more frequently in mixed infections than in any other lineage (V.A. Ellis, personal observation). OZ14 is considered a generalist parasite that has been recovered at a high rate from introduced species such as House Sparrows and European Starlings (Sturnus vulgaris) (R.E. Ricklefs, personal observation).
Discussion
Our survey and analysis of the haemosporidian parasites of birds of eastern Tennessee revealed considerable parasite diversity and complex parasite–host relationships. We found 22 distinct haemosporidian parasite lineages across 25 host species and determined that some variation in parasite prevalence is driven by specific host life history traits; body size, relative abundance, nesting habits, and migratory behavior all appear to contribute to variation in prevalence.
Haemosporidian prevalence did not vary among sites when we examined data by species. This may reflect the relatively small area from which we sampled, as well as our modest sample sizes. In addition, haemosporidian parasite lineages have been shown to respond differently to environmental variables, such as proximity to open water, in the same area (Lachish et al. 2011), which could have erased any site-specific signal in our data. Habitats in our sites were also relatively homogeneous. Our sites were within 80 km of one another, and all were within close proximity to water, which is necessary for reproduction by the dipteran insect vectors (Bates 1949). Since site was not a significant predictor variable, we were able to group our data to analyze potential individual-level and species-level predictors of parasitism.
Host life history traits and infection prevalence
We found no significant relationships between infection status and individual-level traits (e.g., host sex, age, and body condition) in this community, contrary to our original hypothesis. Our results suggest that the individual-level traits examined in this study may not be important predictors of haemosporidian infection across communities. However, we found several patterns in species-level traits indicating that aspects of avian life history and ecology shape, to a limited extent, their parasite community and the proportion of individuals infected by parasites.
Abundance was positively related to total infection prevalence across species, consistent with results from Hochachka and Dhondt (2000) and Brown et al. (2001); this could be a result of the facilitated transmission of parasites with increased host abundance (Dobson 2004). These findings differ from those of Zhang et al. (2014), who found no relationship, and from those of Ricklefs et al. (2005), which revealed a U-shaped relationship between host abundance and parasite prevalence, where the most abundant and least abundant host species had the highest infection prevalence. This U-shaped pattern could be due to high parasite transmission rates in abundant species, and either high parasite virulence or poor host immunity in less abundant species (Ricklefs et al. 2005). We cannot determine the mechanism for the pattern we uncovered or evaluate whether this relationship is likely to be broadly observed, but we speculate that the pattern is driven by vector exposure and feeding rates. We do not have sufficient data to examine patterns of specialization, but it is plausible that parasite lineages can specialize on only abundant hosts, and that less common hosts are infected by spillover (i.e., accidental) infections or by generalists (Medeiros et al. 2014).
Species average body mass was positively related to Haemoproteus prevalence, matching results from Ricklefs et al. (2005) and González et al. (2014). Larger-bodied species may provide more surface area for vector feeding (Hamilton and Zuk 1982; Scheuerlein and Ricklefs 2004), and may better tolerate vector bites, since the amount of blood extracted is relatively small compared to total volume (Hamilton and Zuk 1982), and produce more carbon dioxide, which may attract dipteran vectors (Scheuerlein and Ricklefs 2004).
We hypothesized that some ecological species-level traits, such as foraging stratum and nesting height, might influence haemosporidian prevalence as a result of differential exposure to vectors. Although we found no relationship between any measure of prevalence and foraging level, Svensson-Coelho et al. (2013) found that prevalence of Plasmodium was lowest for Ecuadorian species that forage solely on the ground. In contrast, Astudillo et al. (2013) found that Plasmodium prevalence in Georgia (USA) was highest for middle- to high-level foragers and that Haemoproteus was highest for low- to middle-level foragers. Similar patterns for Haemoproteus were found in Colombia (González et al. 2014). Such conflicting results may arise from differences in sampling seasons, habitat types, host species, or geographic regions, or might reflect type I or type II statistical errors.
Haemosporidian prevalence varied in relation to species nesting height. Haemoproteus prevalence was higher for species that nest between 1 and 5 m above ground level than for species that nest below 1 m or above 5 m. Because canopy heights in our early-successional habitats were low, this pattern is consistent with the observation that Haemoproteus vectors are more abundant in forest canopies (Garvin and Greiner 2003; Cerný et al. 2011; Lassen et al. 2012). Other studies have found similar positive associations between nesting height and parasite prevalence (Fecchio et al. 2011; González et al. 2014; Lutz et al. 2015), while still others have failed to find an association (Ricklefs et al. 2005; Fecchio et al. 2013). We found a non-significant trend of increasing Plasmodium prevalence at lower nesting heights, consistent with the observation of Cerný et al. (2011) that vectors of Plasmodium were more abundant at ground level. The complex and inconsistent patterns that have been observed between nesting or foraging levels and infection prevalence are likely related to vector ecology and habitat use, but clearly require additional study. Although vector sampling was beyond the scope of this study, integrating vector sampling (Medeiros et al. 2013) in future research might resolve these conflicting patterns, across studies, between nesting and foraging strata and haemosporidian prevalence.
We found that total prevalence and prevalence of Plasmodium were higher for species with open-cup nests. Increased infection prevalence in open-cup nests indicates that Plasmodium vectors—culicid mosquitoes—are more likely to come in contact with species that have open-cup nests than with cavity nesters, perhaps because there is no physical barrier between the vectors and the host. However, Fecchio et al. (2011), using microscopy to analyze blood smears, found that closed-cup-nesting birds of the Brazilian Cerrado had higher prevalence of Plasmodium, perhaps due to higher concentrations of carbon dioxide, an olfactory cue used by mosquitoes (Gibson and Torr 1999) in closed-cup nests. Lutz et al. (2015) obtained results similar to those of Fecchio et al. (2011) in their investigation of birds in Malawi using PCR methods. The conflicting results regarding nest type will require further investigation and comparison of a variety of host species with varying nesting strategies.
Avian haemosporidian parasites have depressed the survival of parasite-naïve native populations in Hawaii (van Riper et al. 1986), local populations of European passerine birds (Marzal et al. 2008; Lachish et al. 2011), and individual Blue Tits (Cyanistes caeruleus) in Spain (Martínez-de la Puente et al. 2010). In our study, we found no relationship among species between average annual survival and prevalence of haemosporidian infections. However, if haemosporidian prevalence were temporally dynamic (Fallon et al. 2004), we might expect only periodic changes in host survival, which might not be captured by an average annual survival estimate. Furthermore, particular parasite lineages may have varying degrees of virulence (Lachish et al. 2011), which would also mask a relationship between prevalence of many lineages together and average annual survival.
We found that total prevalence was negatively related to incubation period. Ricklefs (1992) hypothesized that this relationship could result from improved immune function in species with slower embryonic development, and found the same pattern among altricial birds across a broad range of avian families. Diurnal birds of prey also showed a negative relationship (Tella et al. 1999), but a local bird community in Brazil did not (Fecchio et al. 2013).
Solitary species and flocking species did not differ with respect to haemosporidian prevalence. Studies examining the influence of flocking behavior on haemosporidian prevalence have produced mixed results. Lutz et al. (2015) found that the prevalence of Haemoproteus and Plasmodium was higher in species that form single-species flocks than in species in which individuals remain solitary or join mixed-species flocks. However, González et al. (2014) found that the prevalence of Haemoproteus was higher in species forming mixed-species flocks than in those that do not, although overall infection prevalence was low (16 %) in this microscopy-based study. Because haemosporidian parasites shift between closely related species more than expected by chance, the influence of mixed-species flocking might depend on the phylogenetic relatedness of participating species, the degree of parasite specialization, and other host traits that influence differential exposure to vectors.
The prevalence of Haemoproteus was lower in neotropical migrants than in short-distance migrants or residents. We had expected higher prevalence in neotropical migrants because, during their annual cycle, they are exposed to more vector and parasite species (Waldenström et al. 2002) and spend more time in regions where vectors are actively feeding. We found no relationship with respect to migration status for prevalence of Plasmodium or total prevalence, and Fecchio et al. (2013) found no relationship between prevalence and migration. Our finding for Haemoproteus is difficult to explain. Of our three most commonly sampled neotropical migrants (Indigo Bunting Passerina cyanea, Common Yellowthroat Geothlypis trichas, and Yellow-breasted Chat Icteria virens), only one of 111 individuals was infected with Haemoproteus, while 61 were infected with Plasmodium. Our results might reflect the idiosyncracies of these three species, which may not be representative of neotropical migrants.
The parasite-mediated sexual selection hypothesis suggests that sexual dimorphism may exist as a result of the evolution of energetically costly signals by males to demonstrate their resistance to pathogens (Hamilton and Zuk 1982). However, Hamilton and Zuk (1982) and subsequent studies (Doucet and Montgomerie 2003; Scheuerlein and Ricklefs 2004) used blood smears to measure parasite prevalence, which consistently underestimate prevalence measured with molecular techniques (Fallon et al. 2003; Fallon and Ricklefs 2008), and even molecular techniques can miss low-level infections and underestimate true prevalence (Valkiūnas et al. 2008). Our PCR-based estimates of parasite prevalence did not reveal any significant differences with respect to degree of sexual dimorphism, consistent with Ricklefs et al. (2005). Nevertheless, we did not incorporate information on UV-reflective plumage in the determination of avian sexual dimorphism in our analysis, and so our results should be interpreted cautiously. To date, few studies have incorporated UV-reflective plumage to categorize sexual dimorphism (Eaton 2005, but see Garamszegi and Møller 2012), and this should be addressed in future studies of parasite prevalence and host traits.
Introduced species can lose native parasites, and often harbor fewer parasite species than do native hosts (Torchin et al. 2003); House Sparrows conform to this pattern, and exhibit lower diversity and prevalence of haemosporidian parasites in colonized regions (Marzal et al. 2011). In our study, only one in 12 (8 %) House Sparrows was infected, and this was significantly lower than the mean prevalence across all species (χ 2 = 4.59; df = 1; p = 0.032). The infected bird had a mixed infection, with both Plasmodium (OZ01) and Haemoproteus (CHI20PA). Both lineages were common generalists in our study and elsewhere in North and South America (Ricklefs et al. 2014), and their occurrence in House Sparrows accords with Marzal et al. (2011), who found that House Sparrows in their introduced range harbored local generalist parasites.
A relevant regional comparison
Two parasite lineages (OZ01 and OZ03) showed marked differences in prevalence between our survey and that of Ricklefs et al. (2005) in the Ozarks. OZ01, a common host-generalist, was the most frequent lineage in eastern Tennessee, and was the third most frequent in the Ozarks. OZ01 and OZ03 accounted for 38 and 22 % of all infections in east Tennessee, but only 12 and 5 %, respectively, in the Ozarks. These differences in haemosporidian communities could result from habitat or climatic differences between eastern Tennessee and the Ozarks in southern Missouri, or differences in their avian communities, or they could reflect stochastic community processes. In contrast, OZ08 was found predominately in Yellow-breasted Chats in both eastern Tennessee (16 of 17 infections; 94 %) and the Ozarks (23 of 25 infections; 92 %), and it occurs in breeding populations of the chat on the Yucatan Peninsula of southern Mexico (R.E. Ricklefs, unpublished data), illustrating that the lineage is not restricted geographically and appears to specialize on the same host across its range. Comparing our results to those of Ricklefs et al. (2005) highlights the complexity of these parasite-host associations and how these communities can change over short spatial scales (Hellgren et al. 2009), warranting continued finer-scale investigations across and within regions.
Multiple infections
We detected multiple infections in 20 individuals (14 %) from ten species (40 %) of our parasitized samples (Table 4). With an overall minimum infection prevalence of 44 % in our system, we would expect at least 0.442 (19 %), or approximately 27 individuals, to have multiple infections by chance alone. Because the prevalence of multiple infections was somewhat lower than expected, and because most lineages are not host-specific, it would seem either that detecting multiple infections is difficult, or that some parasite lineages prevent coinfection, possibly through cross-immunity by the host. Fecchio et al. (2013) found a similar multiple infection rate of 17 %, and Astudillo et al. (2013) found multiple infections in ~28 % of individuals in three of their four targeted species that were infected with haemoparasites. Several methods have been proposed to screen for multiple infections (Beadell and Fleischer 2005; Pérez-Tris and Bensch 2005; Valkiūnas et al. 2006; Lutz et al. 2015). We suggest that future studies report the prevalence of mixed infections and attempt to partition them out, perhaps by using the phasing method that we describe in Online Resource 1, to contribute to the growing knowledge of malaria parasites and host interactions.
Conclusions
We examined factors that influence haemosporidian parasitism rates within and across avian species. Our analyses emphasize the complexity of host–parasite interactions. Parasite prevalence varied across species, and some species-level traits, including abundance and nesting height, were associated with parasite prevalence. We suggest that these population and host traits result in differential exposure to infective vectors, and thus infection prevalence. Research on habitat use of vectors (e.g., vertical stratification in the canopy) might explain inconsistent results across studies and elucidate other factors that influence prevalence. Intrinsic traits of host species, such as resistance to pathogens or incubation period (Ricklefs 1992), and intrinsic traits of parasite lineages, including those that determine host specialization, also influence prevalence. In addition, some parasite–host associations and the prevalence of parasite lineages varied spatially. The avian haemosporidian parasite–host community in eastern Tennessee adds to our understanding of the distribution and diversity of avian haemosporidian parasites, examines the ecological factors that influence host susceptibility, and demonstrates the complex, spatially variable nature of this parasite–host system.
References
Astudillo VG, Hernández SM, Kistler WM et al (2013) Spatial, temporal, molecular, and intraspecific differences of haemoparasite infection and relevant selected physiological parameters of wild birds in Georgia, USA. Int J Parasitol Parasites Wildl 2:178–189. doi:10.1016/j.ijppaw.2013.04.005
Bates M (1949) The natural history of mosquitoes. Macmillan, New York
Beadell JS, Fleischer RC (2005) A restriction enzyme-based assay to distinguish between avian hemosporidians. J Parasitol 91:683–685. doi:10.1645/GE-3412RN
Bensch S, Waldenström J, Jonzén N et al (2007) Temporal dynamics and diversity of avian malaria parasites in a single host species. J Anim Ecol 76:112–122. doi:10.2307/4125100
Bivand RS, Pebesma E, Gómez-Rubio V (2013) Applied spatial data analysis with R. Springer, New York
Borgia G, Collis K (1989) Female choice for parasite-free male satin bowerbirds and the evolution of bright male plumage. Behav Ecol Sociobiol 25:445–453. doi:10.1007/BF00300191
Brown CR, Brown MB (1992) Ectoparasitism as a cause of natal dispersal in cliff swallows. Ecology 73:1718–1723. doi:10.2307/1940023
Brown CR, Komar N, Quick SB et al (2001) Arbovirus infection increases with group size. Proc R Soc Lond Ser B Biol Sci 268:1833–1840. doi:10.1098/rspb.2001.1749
Cerný O, Votýpka J, Svobodová M (2011) Spatial feeding preferences of ornithophilic mosquitoes, blackflies and biting midges. Med Vet Entomol 25:104–108. doi:10.1111/j.1365-2915.2010.00875.x
Dawson RD, Bortolotti GR (2000) Effects of hematozoan parasites on condition and return rates of American kestrels. Auk 117:373–380. doi:10.2307/4089719
Desante DF, Burton KM, Saracco JF, Walker BL (1995) Productivity indices and survival rate estimates from MAPS, a continent-wide programme of constant-effort mist-netting in North America. J Appl Stat 22:935–948. doi:10.1080/02664769524720
Dobson A (2004) Population dynamics of pathogens with multiple host species. Am Nat 164:S64–S78. doi:10.1086/424681
Doucet SM, Montgomerie R (2003) Structural plumage colour and parasites in satin bowerbirds Ptilonorhynchus violaceus: implications for sexual selection. J Avian Biol 34:237–242. doi:10.1034/j.1600-048X.2003.03113.x
Dunning JB Jr (2008) CRC handbook of avian body masses, 2nd edn. CRC Press, Boca Raton
Eaton MD (2005) Human vision fails to distinguish widespread sexual dichromatism among sexually “monochromatic” birds. Proc Natl Acad Sci USA 102:10942–10946
Fallon SM, Ricklefs RE (2008) Parasitemia in PCR-detected Plasmodium and Haemoproteus infections in birds. J Avian Biol 39:514–522. doi:10.1111/j.2008.0908-8857.04308.x
Fallon SM, Ricklefs RE, Swanson BL, Bermingham E (2003) Detecting avian malaria: an improved polymerase chain reaction diagnostic. J Parasitol 89:1044–1047. doi:10.1645/GE-3157
Fallon SM, Ricklefs RE, Latta SC, Bermingham E (2004) Temporal stability of insular avian malarial parasite communities. Proc R Soc B Biol Sci 271:493–500. doi:10.1098/rspb.2003.2621
Fallon SM, Fleischer RC, Graves GR (2006) Malarial parasites as geographical markers in migratory birds? Biol Lett 2:213–216. doi:10.1098/rsbl.2005.0429
Fecchio A, Lima MR, Silveira P et al (2011) High prevalence of blood parasites in social birds from a neotropical savanna in Brazil. Emu 111:132–138. doi:10.1071/MU10063
Fecchio A, Lima MR, Svensson-Coelho M et al (2013) Structure and organization of an avian haemosporidian assemblage in a Neotropical savanna in Brazil. Parasitology 140:181–192. doi:10.1017/S0031182012001412
Ferrell ST, Snowden K, Marlar AB et al (2007) Fatal hemoprotozoal infections in multiple avian species in a zoological park. J Zoo Wildl Med 38:309–316. doi:10.1638/1042-7260(2007)038[0309:FHIIMA]2.0.CO;2
Fitze PS, Tschirren B, Richner H (2004) Life history and fitness consequences of ectoparasites. J Anim Ecol 73:216–226. doi:10.1111/j.0021-8790.2004.00799.x
Garamszegi LM, Møller AP (2012) The interspecific relationship between prevalence of blood parasites and sexual traits in birds when considering recent methodological advancements. Behav Ecol Sociobiol 66:107–119. doi:10.1007/s00265-011-1259-2
Garvin MC, Greiner EC (2003) Ecology of Culicoides (Diptera: Ceratopogonidae) in southcentral Florida and experimental Culicoides vectors of the avian hematozoan Haemoproteus danilewskyi Kruse. J Wildl Dis 39:170–178. doi:10.7589/0090-3558-39.1.170
Garvin MC, Remsen JV, Bishop MA, Bennett GF (1993) Hematozoa from passeriform birds in Louisiana. J Parasitol 79:318–321. doi:10.2307/3283564
Gibson G, Torr SJ (1999) Visual and olfactory responses of haematophagous Diptera to host stimuli. Med Vet Entomol 13:2–23. doi:10.1046/j.1365-2915.1999.00163.x
González AD, Matta NE, Ellis VA et al (2014) Mixed species flock, nest height, and elevation partially explain avian haemoparasite prevalence in Colombia. PLoS One 9:e100695. doi:10.1371/journal.pone.0100695
Hamilton WD, Zuk M (1982) Heritable true fitness and bright birds: a role for parasites? Science 218:384–387. doi:10.1126/science.7123238
Hellgren O, Pérez-Tris J, Bensch S (2009) A jack-of-all-trades and still a master of some: prevalence and host range in avian malaria and related blood parasites. Ecology 90:2840–2849. doi:10.1890/08-1059.1
Hijmans RJ, van Etten J, Cheng J et al (2014) raster: geographic data analysis and modeling. R package version 2.1-16. http://CRAN.R-project.org/package=raster
Hochachka WM, Dhondt AA (2000) Density-dependent decline of host abundance resulting from a new infectious disease. Proc Natl Acad Sci USA 97:5303–5306. doi:10.1073/pnas.080551197
Hudson PJ, Dobson AP, Lafferty KD (2006) Is a healthy ecosystem one that is rich in parasites? Trends Ecol Evol 21:381–385. doi:10.1016/j.tree.2006.04.007
Isaaks EH, Srivastava RM (1989) Applied geostatistics. Oxford University Press, New York
Isaksson C, Sepil I, Baramidze V, Sheldon BC (2013) Explaining variance of avian malaria infection in the wild: the importance of host density, habitat, individual life-history and oxidative stress. BMC Ecol 13:1–11. doi:10.1186/1472-6785-13-15
Knowles SCL, Palinauskas V, Sheldon BC (2010) Chronic malaria infections increase family inequalities and reduce parental fitness: experimental evidence from a wild bird population. J Evol Biol 23:557–569. doi:10.1111/j.1420-9101.2009.01920.x
Kuris AM, Hechinger RF, Shaw JC et al (2008) Ecosystem energetic implications of parasite and free-living biomass in three estuaries. Nature 454:515–518. doi:10.1038/nature06970
Lachish S, Knowles SCL, Alves R et al (2011) Fitness effects of endemic malaria infections in a wild bird population: the importance of ecological structure. J Anim Ecol 80:1196–1206. doi:10.1111/j.1365-2656.2011.01836.x
Lafferty KD, Dobson AP, Kuris AM (2006) Parasites dominate food web links. Proc Natl Acad Sci USA 103:11211–11216. doi:10.1073/pnas.0604755103
Lafferty KD, Allesina S, Arim M et al (2008) Parasites in food webs: the ultimate missing links. Ecol Lett 11:533–546. doi:10.1111/j.1461-0248.2008.01174.x
Lassen SB, Nielsen SA, Kristensen M (2012) Identity and diversity of blood meal hosts of biting midges (Diptera: Ceratopogonidae: Culicoides Latreille) in Denmark. Parasit Vectors 5:143. doi:10.1186/1756-3305-5-143
Loiseau C, Iezhova T, Valkiūnas G et al (2010) Spatial variation of haemosporidian parasite infection in African rainforest bird species. J Parasitol 96:21–29. doi:10.1645/GE-2123.1
Longmire JL, Maltbie M, Baker RJ (1997) Use of “‘lysis buffer’” in DNA isolation and its implication for museum collections. Occas Pap Mus Tex Tech Univ 163:1–4
Lutz HL, Hochachka WM, Engel JI et al (2015) Parasite prevalence corresponds to host life history in a diverse assemblage of afrotropical birds and haemosporidian parasites. PLoS One 10:e0121254. doi:10.1371/journal.pone.0121254
Martínez-de la Puente J, Merino S, Tomás G et al (2010) The blood parasite Haemoproteus reduces survival in a wild bird: a medication experiment. Biol Lett 6:663–665. doi:10.1098/rsbl.2010.0046
Marzal A, De Lope F, Navarro C, Møller AP (2005) Malarial parasites decrease reproductive success: an experimental study in a passerine bird. Oecologia 142:541–545. doi:10.1007/s00442-004-1757-2
Marzal A, Bensch S, Reviriego M et al (2008) Effects of malaria double infection in birds: one plus one is not two. J Evol Biol 21:979–987. doi:10.1111/j.1420-9101.2008.01545.x
Marzal A, Ricklefs RE, Valkiūnas G et al (2011) Diversity, loss, and gain of malaria parasites in a globally invasive bird. PLoS One 6:1–8. doi:10.1371/journal.pone.0021905
Medeiros MJ, Eiben JA, Haines WP et al (2013) The importance of insect monitoring to conservation actions in Hawaii. Hawaii Entomol Soc 45:149–166
Medeiros MCI, Ellis VA, Ricklefs RE (2014) Specialized avian Haemosporida trade reduced host breadth for increased prevalence. J Evol Biol 27:2520–2528. doi:10.1111/jeb.12514
Michel N, DeSante DF, Kaschube DR et al (2011) The Monitoring Avian Productivity and Survivorship (MAPS) Program annual reports, 1989–2006. NBII/MAPS Avian Demographics Query Interface. http://www.birdpop.org/nbii2006/NBIIHome.asp
Møller AP (2008) Flight distance and blood parasites in birds. Behav Ecol 19:1305–1313. doi:10.1093/beheco/arn074
Norris K, Anwar M, Read AF (1994) Reproductive effort influences the prevalence of haematozoan parasites in great tits. J Anim Ecol 63:601–610. doi:10.2307/5226
Norte AC, Araújo PM, Sampaio HL et al (2009) Haematozoa infections in a Great Tit Parus major population in Central Portugal: relationships with breeding effort and health. Ibis 151:677–688. doi:10.1111/j.1474-919X.2009.00960.x
Olival KJ, Stiner EO, Perkins SL (2007) Detection of Hepatocystis sp. in southeast Asian flying foxes (Pteropodidae) using microscopic and molecular methods. J Parasitol 93:1538–1540. doi:10.1645/GE-1208.1
Pérez-Tris J, Bensch S (2005) Diagnosing genetically diverse avian malarial infections using mixed-sequence analysis and TA-cloning. Parasitology 131:15–23. doi:10.1017/S003118200500733X
Pérez-Tris J, Hasselquist D, Hellgren O et al (2005) What are malaria parasites? Trends Parasitol 21:209–211. doi:10.1016/j.pt.2005.03.007
Perkins SL, Schall JJ (2002) A molecular phylogeny of malarial parasites recovered from cytochrome b gene sequences. J Parasitol 88:972–978. doi:10.1645/0022-3395(2002)088[0972:AMPOMP]2.0.CO;2
Pyle P (1997) Identification guide to North American birds: a compendium of information on identifying, ageing, and sexing “near-passerines” and passerines in the hand. Slate Creek Press, Bolinas
Read AF (1987) Comparative evidence supports the Hamilton and Zuk hypothesis on parasites and sexual selection. Nature 328:68–70. doi:10.1038/328068a0
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Ribeiro SF, Sebaio F, Branquinho FCS et al (2005) Avian malaria in Brazilian passerine birds: parasitism detected by nested PCR using DNA from stained blood smears. Parasitology 130:261–267. doi:10.1017/S0031182004006596
Richner H, Christe P, Oppliger A (1995) Paternal investment affects prevalence of malaria. Proc Natl Acad Sci USA 92:1192–1194. doi:10.1073/pnas.92.4.1192
Ricklefs RE (1992) Embryonic development period and the prevalence of avian blood parasites. Proc Natl Acad Sci USA 89:4722–4725. doi:10.1073/pnas.89.10.4722
Ricklefs RE, Swanson BL, Fallon SM et al (2005) Community relationships of avian malaria parasites in southern Missouri. Ecol Monogr 75:543–559. doi:10.2307/4539117
Ricklefs RE, Outlaw DC, Svensson-Coelho M et al (2014) Species formation by host shifting in avian malaria parasites. Proc Natl Acad Sci USA 111:14816–14821. doi:10.1073/pnas.1416356111
Rodewald P (ed) (2015) The Birds of North America Online. Cornell Laboratory of Ornithology, Ithaca, NY. http://bna.birds.cornell.edu/BNA/
SAS Institute Inc. (2011) Base SAS® 9.3 procedures guide. SAS Institute Inc, Cary
Sauer JR, Hines JE, Fallon JE et al (2011) The North American breeding bird survey, results and analysis 1966–2010. Version 12.07.2011. USGS Patuxent Wildlife Research Center, Laurel
Scheuerlein A, Ricklefs RE (2004) Prevalence of blood parasites in European passeriform birds. Proc R Soc B Biol Sci 271:1363–1370. doi:10.1098/rspb.2004.2726
Schrader MS, Walters EL, James FC, Greiner EC (2003) Seasonal prevalence of a haematozoan parasite of red-bellied woodpeckers (Melanerpes carolinus) and its association with host condition and overwinter survival. Auk 120:130–137. doi:10.1642/0004-8038(2003)120[0130:SPOAHP]2.0.CO;2
Schulte-Hostedde AI, Zinner B, Millar JS, Hickling GJ (2005) Restitution of mass-size residuals: validating body condition indices. Ecology 86:155–163. doi:10.1890/04-0232
Svensson LME, Ricklefs RE (2009) Low diversity and high intra-island variation in prevalence of avian Haemoproteus parasites on Barbados, Lesser Antilles. Parasitology 136:1121–1131. doi:10.1017/S0031182009990497
Svensson-Coelho M, Blake JG, Loiselle BA et al (2013) Diversity, prevalence, and host specificity of avian Plasmodium and Haemoproteus in a Western Amazon assemblage. Ornithol Monogr 76:1–47
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Tella JL, Blanco G, Forero MG et al (1999) Habitat, world geographic range, and embryonic development of hosts explain the prevalence of avian hematozoa at small spatial and phylogenetic scales. Proc Natl Acad Sci USA 96:1785–1789. doi:10.1073/pnas.96.4.1785
Thomas F, Schmidt-Rhaesa A, Martin G et al (2002) Do hairworms (Nematomorpha) manipulate the water seeking behaviour of their terrestrial hosts? J Evol Biol 15:356–361. doi:10.1046/j.1420-9101.2002.00410.x
Torchin ME, Lafferty KD, Dobson AP et al (2003) Introduced species and their missing parasites. Nature 421:628–630. doi:10.1038/nature01346
Valkiūnas G (2005) Avian malaria parasites and other haemosporidia. CRC Press, Boca Raton
Valkiūnas G, Bensch S, Iezhova TA et al (2006) Nested cytochrome b polymerase chain reaction diagnostics underestimate mixed infections of avian blood Haemosporidian parasites: microscopy is still essential. J Parasitol 92:418–422. doi:10.1645/GE-3547RN.1
Valkiūnas G, Iezhova TA, Križanauskienė A et al (2008) A comparative analysis of microscopy and PCR-based detection methods for blood parasites. J Parasitol 94:1395–1401. doi:10.1645/GE-1570.1
van Riper C, van Riper SG, Goff ML, Laird M (1986) The epizootiology and ecological significance of malaria in Hawaiian land birds. Ecol Monogr 56:327–344. doi:10.2307/1942550
Waldenström J, Bensch S, Kiboi S et al (2002) Cross-species infection of blood parasites between resident and migratory songbirds in Africa. Mol Ecol 11:1545–1554
Wilson K, Bjørnstad ON, Dobson AP et al (2002) Heterogeneities in macroparasite infections: patterns and processes. In: Hudson PJ, Rizzoli A, Grenfell BT et al (eds) The ecology of wildlife diseases. Oxford University Press, Oxford, pp 6–44
Zhang Y, Wu Y, Zhang Q et al (2014) Prevalence patterns of avian Plasmodium and Haemoproteus parasites and the influence of host relative abundance in southern China. PLoS One 9:e99501. doi:10.1371/journal.pone.0099501
Zuk M, McKean KA (1996) Sex differences in parasite infections: patterns and processes. Int J Parasitol 26:1009–1023. doi:10.1016/S0020-7519(96)00086-0
Acknowledgments
Bill Smith, Justine Cucchiara, Stephen Lyn Bales, Knox County Parks and Recreation, and Jeff Debree provided access to banding sites. Anna Harris provided useful suggestions on the project design, and Jonathan Fitz Gerald provided assistance with laboratory techniques. Sean Gunter and Sarah Walters generously provided housing accommodations. This work was supported by grants from the National Science Foundation Malaria Research Coordination Network, the Whitney R. Harris World Ecology Center, and the Tennessee Ornithological Society. The experiments in this study comply with the current laws of the United States. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. C. Klasing.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Matthews, A.E., Ellis, V.A., Hanson, A.A. et al. Avian haemosporidian prevalence and its relationship to host life histories in eastern Tennessee. J Ornithol 157, 533–548 (2016). https://doi.org/10.1007/s10336-015-1298-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10336-015-1298-y