
Abstract
Streptomyces lincolnensis is generally utilized for the production of lincomycin A (Lin-A), a clinically useful antibiotic to treat Gram-positive bacterial infections. Three methylation steps, catalyzed by three different S-adenosylmethionine (SAM)-dependent methyltransferases, are required in the biosynthesis of Lin-A, and thus highlight the significance of methyl group supply in lincomycin production. In this study, we demonstrate that externally supplemented SAM cannot be taken in by cells and therefore does not enhance Lin-A production. Furthermore, bioinformatics and in vitro enzymatic assays revealed there exist two SAM synthetase homologs, MetK1 (SLCG_1651) and MetK2 (SLCG_3830) in S. lincolnensis that could convert l-methionine into SAM in the presence of ATP. Even though we attempted to inactivate metK1 and metK2, only metK2 was deleted in S. lincolnensis LCGL, named as ΔmetK2. Following a reduction of the intracellular SAM concentration, ΔmetK2 mutant exhibited a significant decrease of Lin-A in comparison to its parental strain. Individual overexpression of metK1 or metK2 in S. lincolnensis LCGL either elevated the amount of intracellular SAM, concomitant with 15% and 22% increase in Lin-A production, respectively. qRT-PCR assays showed that overexpression of either metK1 or metK2 increased the transcription of lincomycin biosynthetic genes lmbA and lmbR, and regulatory gene lmbU, indicating SAM may also function as a transcriptional activator. When metK1 and metK2 were co-expressed, Lin-A production was increased by 27% in LCGL, while by 17% in a high-yield strain LA219X.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lincomycin A (Lin-A) is widely used for the treatment of Gram-positive bacterial infections [26], mainly produced by the actinomycete Streptomyces lincolnensis. Lin-A consists of an amino sugar precursor α-methylthiolincosaminide (MTL) and an amino acid derivative N-methylated 4-propyl-l-proline (PPL) moiety [34]. Lin-A and its semi-synthetic derivative clindamycin belong to lincosamide family antibiotics, and clindamycin can be used for the treatment of protozoal diseases, e.g., malaria [26]. Given the high clinical importance of lincosamide antibiotics, enhancement of lincomycin production in S. lincolnensis has been performed by genetic manipulation, fermentation engineering or classical mutagenesis methods over the past 50 years [3, 8, 13, 33].
The lincomycin biosynthetic gene cluster in S. lincolnensis spans over 35 kb of DNA, containing 29 genes concerning the biosynthesis, regulation and resistance [11, 19]. In the process of lincomycin biosynthesis, three methylation steps are required for C-, S-, and N-positions, respectively [9, 25]. Previous reports demonstrated that the methyltransferase LmbJ converted N-demethyllincomycin into lincomycin, and LmbW was involved in the propylproline biosynthesis of Lin-A using S-adenosylmethionine (SAM) as a methyl donor [14, 18]. LmbG was predicted to be responsible for the S-methyl reaction [9, 25] (Fig. 1). These investigations suggest that methyl group supply plays a vital role in lincomycin biosynthesis in S. lincolnensis. A number of studies have confirmed that SAM is an important methyl donor in both primary and secondary metabolisms [2, 4], and overexpression of SAM synthetase gene metK or exogenous addition of SAM enhances the production of multiple types of antibiotics in actinomycetes [16, 35, 36]. SAM is also found to activate transcription factors, which in turn regulate antibiotic biosynthesis of Streptomyces [4, 15, 22, 32]. We have fully sequenced the genome of a lincomycin producer S. lincolnensis LC-G, and found that there exist two metK homologs, metK1 (SLCG_1651) and metK2 (SLCG_3830) (Accession number CP022744 in Genbank). Recently, Pang et al. [18] reported that co-overexpression of lmbW and metK increased lincomycin production and purity in the industrial strain S. lincolnensis SyBE2901. Sequence alignment revealed that the reported metK from S. lincolnensis SyBE2901 was identical to metK1 from S. lincolnensis LC-G. Here we report the function of metK2, as well as metK1, in S. lincolnensis, which could be used for enhancing Lin-A production.

Lincomycin structure and the sites methylated by LmbJ, LmbW, and LmbG, respectively
Materials and methods
Strains, plasmids and growth conditions
All strains and plasmids used in this study are listed in Table 1. Escherichia coli was cultured in Luria–Bertani (LB) medium at 37 °C, with shaking at 220 rpm, supplemented with appropriate antibiotics as required [20]. S. lincolnensis and its derivatives were grown at 30 °C with shaking at 220 rpm in liquid TSBY medium (3% tryptone soya broth, 0.5% yeast extract, 10.3% sucrose, with/without apramycin or thiostrepton) for DNA extraction, or on solid MGM medium (2% soluble starch, 0.5% soybean flour, 0.1% KNO3, 0.05% NaCl, 0.05% MgSO4, 0.05% K2HPO4, 0.001% FeSO4, 2% agar, with/without apramycin or thiostrepton) for sporulation. Spores were isolated and stored in 20% glycerol at -80 °C. Liquid SM medium (0.4% yeast extract, 0.4% tryptone soya broth, 1% glucose, 0.005% MgSO4, 0.02% KH2PO4, 0.04% K2HPO4) was used for S. lincolnensis protoplast preparation [6].
Fermentation and HPLC analysis of Lin-A
Streptomyces lincolnensis LC-G and its derivatives were grown on MGM for sporulation (with appropriate antibiotics for the recombinant strains). The spore suspension was inoculated into a 250-ml flask containing 30 ml of the seed medium (2% soluble starch, 1% glucose, 1% soybean flour, 3% cream corn, 0.15% (NH4)2SO4, 0.4% CaCO3 for culture, with/without apramycin) at 30 °C with shaking at 240 rpm for 2 days. 2 ml seed culture was transferred into 30 ml fermentation medium (10% glucose, 2% soybean flour, 0.15% cream corn, 0.8% NaNO3, 0.5% NaCl, 0.6% (NH4)2SO4, 0.03% K2HPO4, 0.8% CaCO3, with/without apramycin). All fermentation cultures were incubated at 30 °C and 240 rpm for 7 days. After fermentation, 200 μl supernatant of fermentation broth was mixed with 800 μl ethanol, and centrifuged at 12,000 rpm for 10 min to remove the residue. Subsequently, lincomycin extracted from these liquid fermentation cultures was quantified by Waters H13CHA 394G UPLC system on Extend-C18 column (5 μm, 150 × 4.6 mm), which was equilibrated with 60% methyl alcohol and 40% 5 mM ammonium acetate (pH 9.0). The products were monitored at 214 nm. An isocratic program was carried out at a flow rate of 0.4 ml/min.
Exogenous addition of SAM to the culture medium
Streptomyces lincolnensis LC-G was incubated at 30 °C, 240 rpm in fermentation medium as described above. SAM was added to the fermentation medium of S. lincolnensis LC-G after 12 h inoculation, in a final concentration of 0, 0.1, 0.5, 1 and 2 mM, respectively.
Cloning, expression and purification of MetK1 and MetK2 in E. coli
Two DNA fragments encoding MetK1 and MetK2 from LC-G were obtained by PCR using the primers in Table S1. The PCR products were digested with NdeI/EcoRI restriction enzymes and inserted into the corresponding sites of pET-28a (Novagen), generating N-terminal His6-tag fusions. The constructed plasmids pET28a-MetK1 and pET28a-MetK2 were, respectively, introduced into E. coli BL21 (DE3), and the expression of the two proteins was induced with IPTG at a final concentration of 0.5 mM at 16 °C for 20 h. MetK1 and MetK2 His6-tagged proteins were extracted and purified on an Ni2+–NTA spin column (BIO-RAD). The concentrations of the purified proteins were quantified by BCA assays, and their purity was judged by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis.
Enzymatic activity assay of MetK1 and MetK2
The activities of MetK1 and MetK2 were assayed as reported by Oh et al. [15]. Briefly, either MetK1 or MetK2 protein was incubated for 2 h at 37 °C in a reaction mixture (100 μl) containing 0.1 mM Tris/HCl (pH 8.2), 5 mM ATP, 5 mM l-methionine, 200 mM KCl and 10 mM MgCl2. The controls were performed without MetK1 and MetK2, or without ATP and l-methionine. All reactions were terminated by immediately placing the reaction vessels in an ice-water mixture. The terminated reaction mixtures were analyzed by Waters H13CHA 394G UPLC system on Extend-C18 column (5 μm, 250 × 4.6 mm), which was equilibrated with 100 mM phosphate buffer at pH 6.8 (A phase) and methanol (B phase) at a ratio of 80:20 (v/v). The products were monitored at 254 nm. An isocratic program was carried out at a flow rate of 0.5 ml/min. Furthermore, kinetic parameters were determined with the constant concentration of ATP (10 mM). The concentration of l-methionine varied between 0 and 5 mM. Velocity was plotted as a function of varied substrate concentration and the data were fitted to the Michaelis–Menten equation to calculate values of Km and Vmax [5].
Determination of intracellular/extracellular SAM concentration
Intracellular and extracellular SAM levels were determined as described by Oh et al. [15]. During the fermentation of S. lincolnensis LC-G and its derivatives with or without additional SAM (2, 4 and 6 days), 1 ml of the fermentation broth was centrifuged at 5,000 rpm for 10 min. The supernatant was directly applied on UPLC to determine the extracellular SAM concentration. After removal of the supernatant, the intracellular SAM was extracted with 0.5 ml of 1 M formic acid at 4 °C for 1 h. After centrifugation at 12,000 rpm for 15 min, the SAM extracted was quantified by UPLC with the same method above.
Construction of S. lincolnensis LCGL
In order to use the ΦC31-based integrative vector pIB139 [31], a 240-bp DNA fragment was synthesized at Sangon Biotech (Shanghai) Co., Ltd, containing four tandem ΦC31 attB sites [30]. The sequence was as follows: AAAGAATTCCTTCTCTCTAGACGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTAACTAGTGGATCTCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTAACTAGTGGATCTCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTAACTAGTGGATCTCGGGTGCCAGGGCGTGCCCTTGGGCTCCCCGGGCGCGTAACTAGTGGATCCCTGGAGAAGCTTAAA (EcoRI/XbaI and BamHI/HindIII restriction sites were shown in italics, and four attB sequences were underlined). With S. lincolnensis LC-G genomic DNA as a template, two 2.0-kb DNA fragments flanking SLCG_7011 putatively encoding a nonribosomal peptide synthetase (NRPS) were amplified by PCR using two primer pairs attB-P1/attB-P2 and attB-P3/attB-P4 (Table S1). The PCR products and the synthetic 4 × attBΦC31 were, respectively, digested with EcoRI/KpnI, XbaI/HindIII and XbaI/KpnI, and then ligated into the corresponding sites of pKC1139, obtaining pKC1139-4 × attBΦC31. By PEG3350-mediated protoplast transformation, pKC1139-4 × attBΦC31 was introduced into LC-G. After two round homologous chromosomic recombination, the desired mutant with 4 × attBΦC31 instead of SLCG_7011, named as S. lincolnensis LCGL, was obtained and further confirmed by PCR analysis using the primers attB-P5/attB-P6 (Table S1).
In accordance with above procedures, we constructed S. lincolnensis LA219X containing 4 × attBΦC31 from a high-yield S. lincolnensis LA219.
Inactivation, complementation and overexpression of metK1 and metK2 in S. lincolnensis
We tried many methods to disrupt metK1 gene in both S. lincolnensis LC-G and LCGL, but failed, while it was very easy to obtain ΔmetK2 mutants.
The plasmid pKC1139-ΔmetK2, with an internal 717-bp deletion of metK2, was constructed in two steps. Firstly, with the genomic DNA of LC-G as a template, two 1.5-kb fragments flanking metK2 were, respectively, amplified by PCR using two primer pairs metK2-P1/metK2-P2 and metK2-P3/metK2-P4 (Table S1), cleaved by HindIII/XbaI and KpnI/EcoRI, and ligated into the corresponding sites of pUCTSR [7], yielding pUCTSR-ΔmetK2. Secondly, 4.5 kb DNA fragment was digested with EcoRI/HindIII from pUCTSR-ΔmetK2, and then cloned into the same site of pKC1139 [1], generating pKC1139-ΔmetK2. By PEG3350-mediated protoplast transformation, pKC1139-ΔmetK2 was introduced into LCGL. By homologous chromosomic recombination, a 717-bp fragment of the metK2 gene was replaced by thiostrepton resistance gene (tsr) in S. lincolnensis LCGL. The desired ΔmetK2 mutant was further confirmed by PCR amplification using the primers metK2-P5 and metK2-P6 (Table S1). For the complementation of metK2 in ΔmetK2 mutant, a 1221-bp metK2 was amplified with the primers metK2-P7 and metK2-P8 (Table S1), cleaved with NdeI/XbaI, and ligated into the corresponding sites of pIB139, generating pIB139-metK2. Then, pIB139 and pIB139-metK2 were individually introduced into ΔmetK2 mutant, and the corresponding apramycin-resistant ΔmetK2/pIB139 and ΔmetK2/pIB139-metK2 were obtained and further confirmed by PCR amplification with the primers apr-P1 and apr-P2, respectively (Table S1).
Meanwhile, a 1209-bp metK1 was amplified with the primers metK1-P1 and metK1-P2 (Table S1), digested with NdeI/XbaI, and ligated into the corresponding sites of pIB139, generating pIB139-metK1. pIB139-metK1 and pIB139-metK2 were introduced into LCGL to overexpress metK1 and metK2, respectively.
For co-overexpressing metK1 and metK2, the metK2 fragment with promoter PermE* was amplified from pIB139-metK2 using primers metK2-P9 and metK2-P10 (Table S1), cut with NotI/EcoRV and ligated to the corresponding site of pIB139-metK1, generating pIB139-metK1-metK2. Then the co-expression vector pIB139-metK1-metK2 was introduced into LCGL and LA219X to generate the strains LCGL/pIB139-metK1-metK2 and LA219X/pIB139-metK1-metK2, respectively.
RNA isolation and quantitative real-time PCR assay (qRT-PCR)
The relative transcriptional levels of lmbA, lmbR and lmbU were determined by qRT-PCR analysis. Specific primers were designed as listed in Table S1. Total RNA was isolated from S. lincolnensis LCGL and its derivatives after 24 h grown in fermentation liquid medium using the RNA extraction/purification kit (SBS), and the RNA concentration was determined using a microplate reader (BioTek). Isolated RNA (500 ng) was treated with DNase I (MBI Fermentas), and then reversed using a cDNA synthesis kit (MBI Fermentas). qRT-PCR was performed on the Applied Biosystems QuantStudio 6 Flex system with Maxima™ SYBR Green/ROX qPCR Master Mix (MBI Fermentas). The rpoD gene in S. lincolnensis was used as an internal control, and relative transcription was quantified using a comparative cycle threshold method [12].
Results
SAM supplementation of the culture medium cannot increase Lin-A production
Since SAM is the principal methyl donor in microbial cells, we initially examined the effect of exogenous addition of SAM on lincomycin production. Under the final concentrations of SAM from 0.1 mM to 2 mM in the culture medium, the Lin-A production of LC-G kept unchanged (Fig. 2a). We further found that the extracellular SAM concentration generally remained constant throughout LC-G fermentation (Fig. 2b). Also, the intracellular concentration of SAM did not vary depending on the amount increment of extra added SAM (Fig. 2c). These results suggest that SAM cannot be transported across the membrane of S. lincolnensis cells.

Effect of exogenous SAM addition on Lin-A production in S. lincolnensis LC-G. a Lin-A production after adding SAM with different final concentrations (0, 0.1, 0.5, 1, 2) by UPLC analysis. b Extracellular SAM concentrations under different exogenous addition of SAM after 2, 4 and 6 days incubation. c Intracellular SAM concentrations under different exogenous addition of SAM after 2, 4 and 6 days incubation. Mean values of at least three independent experiments were shown, with the standard deviation indicated by error bars
Both MetK1 and MetK2 enzymatically convert l-methionine into SAM in the presence of ATP
Alignment of the genomic sequences of S. lincolnensis LC-G (Accession number CP022744 in Genbank) and other Streptomyces strains revealed there exist two SAM synthetase genes, SLCG_1651 (metK1) and SLCG_3830 (metK2), with high similarity to other metK genes, such as SCO1476 from S. coelicolor A3(2) (93.5 and 81.5% identities) and SAV_6874 from S. avermitilis MA4680 (93.5 and 83% identities).
To test whether MetK1 and MetK2 from LC-G have SAM synthetase activities, they were successively expressed and purified (Fig. 3a). As shown in the UPLC profiles, both MetK1 and MetK2 could convert l-methionine into SAM in the presence of ATP (Fig. 3b).

Enzymatic analyses of MetK1 and MetK2. a Purification of His6-tagged MetK1 and His6-tagged MetK2. Abbreviations: M, protein molecular weight marker; 1, purified MetK1 protein; 2, purified MetK2 protein. b Both MetK1 and MetK2 converted ATP and l-methionine into SAM. The reaction with either MetK1 or MetK2 was performed in the mixture of ATP, l-methionine, KCl and MgCl2. 1, KCl, MgCl2 and MetK1 or MetK2; 2, ATP, l-methionine, KCl and MgCl2; 3, ATP, l-methionine, KCl, MgCl2 and MetK1; 4, ATP, l-methionine, KCl, MgCl2 and MetK2; 5, standard SAM (retention time = 6.9 min)
Moreover, Michaelis–Menten equation was used to determine the kinetic constants of MetK1 and MetK2, exhibiting the Km and Vmax values of 3.42 mM and 4.12 μmol/L/min for MetK1, and 2.14 mM and 4.88 μmol/L/min for MetK2. MetK2 had a higher catalytic activity than MetK1.
Overexpression of metK1 increases Lin-A production in S. lincolnensis LCGL
Given that the genome of S. lincolnensis LC-G lacks the typical attB site for ΦC31-based pIB139 vector integration, we constructed a new S. lincolnensis LCGL from LC-G by replacing SLCG_7011 with 4 × attBΦC31. No significant differences in cell growth, morphological differentiation and lincomycin production were observed between LCGL and LC-G (Fig. S1).
Because of repeated failures in deleting metK1 gene in LC-G and LCGL, we assume that MetK1 may be essential for LC-G growth. To investigate the role of metK1 gene in the biosynthesis of lincomycin, plasmid pIB139-metK1 was transformed into S. lincolnensis LCGL. Compared with the control LCGL/pIB139, LCGL/pIB139-metK1 successively exhibited 1.8-, 2.5- and 1.8-fold increases in intracellular SAM concentrations during incubation periods of 2, 4 and 6 days (Fig. 4a). Consistent with the increase of intracellular SAM, LCGL/pIB139-metK1 showed a 15% (p < 0.01) higher level of Lin-A production on the seventh day of fermentation than LCGL/pIB139 (Fig. 4b). These results indicate that metK1 overexpression in LCGL leads to enhancement of intracellular SAM concentration and Lin-A production.

Overexpression of metK1 increases the yield of Lin-A in S. lincolnensis LCGL. a Intracellular SAM concentrations of LCGL/pIB139 and LCGL/pIB139-metK1. b Lin-A production of LCGL/pIB139 and LCGL/pIB139-metK1. Mean values of at least three replicates were shown, with the standard deviation indicated by error bars. *p < 0.05, **p < 0.01
Overexpression of metK2 increases Lin-A production in S. lincolnensis LCGL
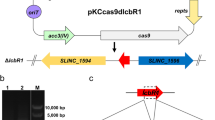
To investigate whether metK2 was also involved in lincomycin biosynthesis, it was disrupted in S. lincolnensis LCGL through tsr replacement, and the resulted mutant ΔmetK2 was confirmed by PCR analysis (Fig. 5a, b). As shown in Fig. 5c, the intracellular SAM concentrations of ΔmetK2 were decreased by approximately 31, 42 and 50% after 2, 4 and 6 days fermentation compared to its parental strain LCGL, respectively (Fig. 5c). Correspondingly, ΔmetK2 had a 55% reduction in Lin-A yield after a 7-day fermentation, from 2.2 to 1.0 g/L (p < 0.001) relative to its parental strain. Complementation of ΔmetK2 with pIB139-metK2 recovered Lin-A production (Fig. 5d). ΔmetK2 showed similar growth rates in YMG liquid medium and sporulation rates on MGM agar medium to its parent strain S. lincolnensis LCGL, indicating that MetK2 is not involved in cell growth and morphological differentiation of S. lincolnensis (Fig. S2).

Effect of inactivation and overexpression of metK2 on Lin-A production in S. lincolnensis LCGL. a Schematic deletion of metK2 in LCGL. b PCR confirmation of ΔmetK2 mutant using the primers metK2-P5 and metK2-P6. A 750-bp band was observed with LCGL, while a 1,500-bp band was detected with ΔmetK2. c Intracellular SAM concentrations in LCGL, ΔmetK2, LCGL/pIB139 and LCGL/pIB139-metK2 cultured for 2, 4 or 6 days. d Lin-A production in LCGL and its derivatives cultured for 7 days. Mean values of at least three independent experiments were shown, with the standard deviation indicated by error bars.*p < 0.05, **p < 0.01, ***p < 0.001
To further probe the involvement of MetK2 in lincomycin biosynthesis, metK2 was overexpressed in LCGL. As shown in Fig. 5c, the levels of the intracellular SAM in LCGL/pIB139-metK2 was, respectively, increased by 2.6-, 3.0- and 2.2-fold during the incubation period of 2, 4 and 6 days in comparison with that of LCGL/pIB139 (Fig. 5c). In accordance with the increased amount of intracellular SAM, the Lin-A yield of LCGL/pIB139-metK2 was increased by 22% (p < 0.001) on the seventh day compared to LCGL/pIB139 (Fig. 5d). These data further confirm that MetK2 is certainly involved in the SAM biosynthesis, further enhancing Lin-A production in S. lincolnensis.
MetK1 and MetK2 promote the transcription of the genes involved in Lin-A biosynthesis and regulation in S. lincolnensis
Since SAM was found to act as an intracellular signal molecule for regulating the antibiotic biosynthesis in Streptomyces independent of its role as a methyl donor [4, 36], we measured the transcripts of lmbA (SLCG_0227, a Lin-A biosynthetic gene encoding gamma-glutamyl transferase), lmbR (SLCG_0245, a Lin-A biosynthetic gene encoding transaldolase) and lmbU (SLCG_0253, a positive regulatory gene controlling Lin-A biosynthesis) by qRT-PCR. The transcriptional levels of lmbA, lmbR and lmbU in LCGL/pIB139-metK1 were, respectively, increased by 1.75-, 1.83- and 2.2-fold at 24 h compared with those in LCGL/pIB139, while the transcriptional levels of lmbA, lmbR and lmbU in metK2 overexpression strain raised by 5.5-, 3.7- and 6.1-fold (Fig. 6). These results demonstrate that SAM surely acts as a signal molecule to promote the transcription of lincomycin biosynthetic genes in S. lincolnensis. Furthermore, MetK2 makes a more significant contribution than MetK1.

Overexpression of metK1 or metK2 enhances the transcription of lmbA, lmbR and lmbU in S. lincolnensis. qRT-PCR was used to quantify the transcriptional levels of lmbA, lmbR and lmbU in LCGL/pIB139, LCGL/pIB139-metK1 and LCGL/pIB139-metK2 cultured for 24 h in fermentation medium. Mean values of at least three independent experiments were shown, with the standard deviation indicated by error bars. *p < 0.05, **p < 0.01
Co-overexpression of metK1 and metK2 further improve Lin-A yield in S. lincolnensis
In order to further improve lincomycin production, metK1 and metK2 were co-overexpressed in the LCGL. The Lin-A yield of LCGL/pIB139-metK1-metK2 was remarkably increased by 27% (p < 0.01) relative to that of LCGL/pIB139 (Fig. 7a).

Co-overexpressing of metK1 and metK2 improves the yield of Lin-A in S. lincolnensis. a Lin-A production of LCGL/pIB139 (control) and LCGL/pIB139-metK1-metK2 (SLCGL-K1-K2). b Lin-A production of LA219X/pIB139 (control) and LA219X/pIB139-metK1-metK2 (SLA219X-K1-K2). Mean values of at least three replicates were shown, with the standard deviation indicated by error bars. **p < 0.01
To examine the applicability and universality of co-overexpressing metK1 and metK2 for enhancement of Lin-A production, the pIB139-metK1-metK2 was introduced into a higher-yield S. lincolnensis strain LA219X. As expected, LA219X/pIB139-metK1-metK2 (2.92 g/L) showed 17% (p < 0.01) improvement of Lin-A production relative to LA129X/pIB139 (2.5 g/L) when cultured in 30 mL industrial fermentation medium for 7 days (Fig. 7b). It is likely that co-overexpressing metK1 and metK2 in other industrial S. lincolnensis strains will be of significantly commercial value.
Discussion
Although the plasmids pPM927 and pSOK804, containing phage VWB and pSAM2 integration sites, have been widely used in the research field of actinomycetes [17, 24, 27, 28], we found that the yields of Lin-A were both lowered by about 50% when they were introduced into S. lincolnensis LC-G (Fig. S1C), indicating that they are not suitable for engineering industrial S. lincolnensis strains. Due to lack of the ΦC31 attB site in the genome of S. lincolnensis LC-G, we used homologous recombination to replace SLCG_7011, putatively encoding a nonribosomal peptide synthetase (NRPS), with ΦC31 attB sites. The resulting mutant LCGL showed similar cell growth, morphological differentiation and lincomycin production relative to LC-G (Fig. S1D, E and F). When pIB139 was introduced into LCGL, there was no effect on the lincomycin production (Fig. S1D), indicating LCGL strain is suitable for plasmids with ΦC31 attB sites to express heterologous genes.
The rationale using attB sequence is that phage ΦC31 integrase catalyzes site-specific exchange to generate stable mutants [23]. For integration, the attB site undergoes the recombination with phage attP site of pSET152-derived plasmid to form hybrid sites attL and attR. Only attL and attR at the franking regions of inserted linear ΦC31-based vector by exconjugants, other attB sites were eliminated [30]. Taken the genomic DNA of S. lincolnensis strains LCGL/pIB139, LCGL/pIB139-metK1, LCGL/pIB139-metK2 and LCGL/pIB139-metK1-metK2 as templates, these DNA fragments between attL and attR were, respectively, amplified with three primer pairs (Fig.S3A, B and C). The presence of attL and attR in above recombinant S. lincolnensis strains was further verified by DNA sequencing (Fig.S4A and B). Thus, our current purpose of inserting the 4× attB cassette with a suitable size of 240 bp is only to facilitate molecular cloning, consistent with the description of using 8x attB cassette in Saccharopolyspora erythraea for site-specific recombination [30].
SAM, acting as a major methyl donor, plays a vital role in primary and secondary metabolism [2]. Most of investigations demonstrated that addition of exogenous SAM or overexpression of SAM synthetase caused common yield improvement of antibiotics in actinomycetes [10, 36]. However, our results showed that extra SAM added to culture media could not enter S. lincolnensis cells. This may be due to lack of a transporter for SAM uptake in S. lincolnensis, which is similar to the phenomenon observed in Streptomyces coelicolor M512 [36].
We have completely sequenced the genome of the lincomycin-producing S. lincolnensis LC-G and found two homologs of SAM synthetase genes, SLCG_1651 (metK1) and SLCG_3830 (metK2). Consistent with other MetKs in Streptomyces [15], both MetK1 and MetK2 harbor a conserved ATP-binding motif and two metal-binding regions for Mg2+ and K+ (Fig. S5A). The metK1 (SLCG_1651) is located among the genes encoding a primosome assembly protein (SLCG_1650), a bifunctional phosphopantothenoylcysteine decarboxylase/phosphopantothenate synthase (SLCG_1652), and an omega subunit of DNA-dependent RNA polymerase (SLCG_1653) (Fig. S5B), inferring MetK1 might be mainly involved in DNA modification and primary metabolism. That may be the reason why we failed to inactivate metK1 in LC-G or LCGL. A similar phenomenon was also reported in Streptomyces peucetius var. caesius [15]. The metK2 (SLCG_3830) is clustered in the metK2-adoK-metH-metF-sahH genes (SLCG_3830 ~ SLCG_3834) (Fig. S3B), which are mostly responsible for the SAM cycle [36]. Pang et al. reported that overexpression of metK could promote the production of Lin-A in an industrial S. lincolnensis SyBE2901 [18]. According to the sequence alignment, we deduced that the metK reported from S. lincolnensis SyBE2901 was identical to the metK1 from S. lincolnensis LC-G. In our study, we found that overexpression of both metK1 and metK2 increased Lin-A production by upgrading the intracellular SAM concentration in S. lincolnensis LCGL, and overexpression of metK2 had a more significant effect. Furthermore, co-overexpression of metK1 and metK2 dramatically enhanced the yield of Lin-A in LCGL, even in a higher lincomycin-producing S. lincolnensis strain. We anticipate that the strategy of co-overexpression of the two SAM synthetase genes will be generally applicable for improvement of other antibiotics in industry.
Previous findings gave a hint that besides a methyl group supplier, SAM also played a role in transcriptional regulation of antibiotic biosynthesis in Streptomyces [15, 22, 32, 36]. Hereby, we provide new evidences that SAM is indeed a signal molecule to regulate the transcription of lincomycin biosynthetic and regulatory genes, but the mechanism remains to be explored.
Change history
30 May 2018
In the online published article, row value “pIB139-metK1-metK2” in table 1 has been processed incorrectly. The correct table is given below
References
Bierman M, Logan R, Obrien K, Seno ET, Rao RN, Schoner BE (1992) Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49. https://doi.org/10.1016/0378-1119(92)90627-2
Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, Mccann PP (1996) S-adenosylmethionine and methylation. FASEB J 10:471–480
Choi D, Cho K (2004) Effect of carbon source consumption rate on lincomycin production from Streptomyces lincolnensis. J Microbiol Biot 14:532–539
Chu J, Qian J, Zhuang Y, Zhang S, Li Y (2013) Progress in the research of S-adenosyl-l-methionine production. Appl Microbiol Biot 97:41–49. https://doi.org/10.1007/s00253-012-4536-8
Copeland RA (2000) Enzymes: a practical introduction to structure, mechanism and data analysis, 2nd edn. Wiley, Hoboken
Du L, Liu R, Ying L, Zhao G (2012) An efficient intergeneric conjugation of DNA from Escherichia coli to mycelia of the lincomycin-producer Streptomyces lincolnensis. Int J Mol Sci 13:4797–4806. https://doi.org/10.3390/ijms13044797
Han S, Song P, Ren T, Huang X, Cao C, Zhang B (2011) Identification of SACE_7040, a member of TetR family related to the morphological differentiation of Saccharopolyspora erythraea. Curr Microbiol 63:121–125. https://doi.org/10.1007/s00284-011-9943-z
Hou B, Wu H, Guo M, Petkovic H, Tao L, Zhu X, Ye J, Zhang H (2018) The novel transcriptional regulator LmbU promotes lincomycin biosynthesis through regulating expression of its target genes in Streptomyces lincolnensis. J Bacteriol 200:e00447–e00417. https://doi.org/10.1128/JB.00447-17
Janata J, Kadlcik S, Koberska M, Ulanova D, Kamenik Z, Novak P, Kopecky J, Novotna J, Radojevic B, Plhackova K (2015) Lincosamide synthetase—a unique condensation system combining elements of nonribosomal peptide synthetase and mycothiol metabolism. PLoS ONE 10:e0118850. https://doi.org/10.1371/journal.pone.0118850
Kim DY, Hwang YI, Choi SU (2011) Cloning of metK from Actinoplanes teichomyceticus ATCC31121 and effect of its high expression on antibiotic production. J Microbiol Biot 21:1294–1298. https://doi.org/10.4014/jmb.1101.01018
Koběrska M, Kopecký J, Olsovska J, Jelinkova M, Ulanova D, Man P, Flieger M, Janata J (2008) Sequence analysis and heterologous expression of the lincomycin biosynthetic cluster of the type strain Streptomyces lincolnensis ATCC 25466. Folia Microbiol 53:395–401. https://doi.org/10.1007/s12223-008-0060-8
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Meng S, Wu H, Wang L, Zhang B, Bai L (2017) Enhancement of antibiotic productions by engineered nitrate utilization in actinomycetes. Appl Microbiol Biot 101:5341–5352. https://doi.org/10.1007/s00253-017-8292-7
Najmanova L, Kutejova E, Kadlec J, Polan M, Olsovska J, Benada O, Novotna J, Kamenik Z, Halada P, Bauer J (2013) Characterization of N-demethyllincosamide methyltransferases LmbJ and CcbJ. ChemBioChem 14:2259–2262. https://doi.org/10.1002/cbic.201300389
Oh T, Niraula NP, Liou K, Sohng J (2010) Identification of the duplicated genes for S-adenosyl-l-methionine synthetase (metK1-sp and metK2-sp) in Streptomyces peucetius var. caesius ATCC 27952. J Appl Microbiol 109:398–407. https://doi.org/10.1111/j.1365-2672.2010.04688.x
Okamoto S, Lezhava A, Hosaka T, Okamotohosoya Y, Ochi K (2003) Enhanced expression of S-adenosylmethionine synthetase causes overproduction of actinorhodin in Streptomyces coelicolor A3(2). J Bacteriol 185:601–609. https://doi.org/10.1128/jb.185.2.601-609.2003
Ostash B, Makitrinskyy R, Walker S, Fedorenko V (2009) Identification and characterization of Streptomyces ghanaensis ATCC14672 integration sites for three actinophage-based plasmids. Plasmid 61:171–175. https://doi.org/10.1016/j.plasmid.2008.12.002
Pang AP, Du L, Lin C, Qiao J, Zhao G (2015) Co-overexpression of lmbW and metK led to increased lincomycin A production and decreased byproduct lincomycin B content in an industrial strain of Streptomyces lincolnensis. J Appl Microbiol 119:1064–1074. https://doi.org/10.1111/jam.12919
Peschke U, Schmidt H, Zhang H, Piepersberg W (1995) Molecular characterization of the lincomycin-production gene cluster of Streptomyces lincolnensis 78-11. Mol Microbiol 16:1137–1156. https://doi.org/10.1111/j.1365-2958.1995.tb02338.x
Sambrook JRD (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, New York
Sekurova ON, Brautaset T, Sletta H, Borgos SEF, Jakobsen OM, Ellingsen TE, Strom AR, Valla S, Zotchev SB (2004) In vivo analysis of the regulatory genes in the Nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455 reveals their differential control over antibiotic biosynthesis. J Bacteriol 186:1345–1354. https://doi.org/10.1128/JB.186.5.1345-1354.2004
Shin S, Xu D, Kwon H, Suh J (2006) S-adenosylmethionine activates adpA transcription and promotes streptomycin biosynthesis in Streptomyces griseus. FEMS Microbiol Lett 259:53–59. https://doi.org/10.1111/j.1574-6968.2006.00246.x
Smith MCM, Brown W, Mcewan AR, Rowley PA (2010) Site-specific recombination by φC31 integrase and other large serine recombinases. Biochem Soc T 38:388–394. https://doi.org/10.1042/BST0380388
Smokvina T, Mazodier P, Boccard F, Thompson CJ, Guerineau M (1990) Construction of a series of pSAM2-based integrative vectors for use in actinomycetes. Gene 94:53–59. https://doi.org/10.1016/0378-1119(90)90467-6
Spizek J, Rezanka T (2017) Lincosamides: chemical structure, biosynthesis, mechanism of action, resistance, and applications. Biochem Pharmacol 133:20–28. https://doi.org/10.1016/j.bcp.2016.12.001
Spižek J, Řezanka T (2004) Lincomycin, clindamycin and their applications. Appl Microbiol Biot 64:455–464
Te Poele EM, Dijkhuizen L (2008) Actinomycete integrative and conjugative elements. Antonie Van Leeuwenhoek 94:127–143. https://doi.org/10.1007/s10482-008-9255-x
Te Poele EM, Oliynyk M, Leadlay PF, Bolhuis H, Dijkhuizen L (2008) Actinomycete integrative and conjugative pMEA-like elements of Amycolatopsis and Saccharopolyspora decoded. Plasmid 59:202–216. https://doi.org/10.1016/j.plasmid.2008.01.003
Wu H, Chen M, Mao Y, Li W, Liu J, Huang X, Zhou Y, Ye B, Zhang L, Weaver DT (2014) Dissecting and engineering of the TetR family regulator SACE_7301 for enhanced erythromycin production in Saccharopolyspora erythraea. Microbial Cell Fact 13:158. https://doi.org/10.1186/s12934-014-0158-4
Wu J, Zhang Q, Deng W, Qian J, Zhang S, Liu W (2011) Toward improvement of erythromycin A production in an industrial Saccharopolyspora erythraea strain via facilitation of genetic manipulation with an artificial attB site for specific recombination. Appl Environ Microbiol 77:7508–7516. https://doi.org/10.1128/AEM.06034-11
Wu P, Pan H, Zhang C, Wu H, Yuan L, Huang X, Zhou Y, Ye B, Weaver DT, Zhang L (2014) SACE_3986, a TetR family transcriptional regulator, negatively controls erythromycin biosynthesis in Saccharopolyspora erythraea. J Ind Microbiol Biot 41:1159–1167. https://doi.org/10.1007/s10295-014-1449-9
Xu D, Kwon H, Suh J (2008) S-adenosylmethionine induces BldH and activates secondary metabolism by involving the TTA-codon control of bldH expression in Streptomyces lividans. Arch Microbiol 189:419–426. https://doi.org/10.1007/s00203-007-0336-4
Ye R, Wang Q, Zhou X (2009) Lincomycin, rational selection of high producing strain and improved fermentation by amino acids supplementation. Bioprocess Biosyst Eng 32:521–529
Zhao Q, Wang M, Xu D, Zhang Q, Liu W (2015) Metabolic coupling of two small-molecule thiols programs the biosynthesis of lincomycin A. Nature 518:115–119. https://doi.org/10.1038/nature14137
Zhao X, Wang Q, Guo W, Cai Y, Wang C, Wang S, Xiang S, Song Y (2013) Overexpression of metK shows different effects on avermectin production in various Streptomyces avermitilis strains. World J Microbiol Biotechnol 29:1869–1875. https://doi.org/10.1007/s11274-013-1350-0
Zhao XQ, Gust B, Heide L (2010) S-adenosylmethionine (SAM) and antibiotic biosynthesis: effect of external addition of SAM and of overexpression of SAM biosynthesis genes on novobiocin production in Streptomyces. Arch Microbiol 192:289–297. https://doi.org/10.1007/s00203-010-0548-x
Acknowledgements
We are grateful to Dr. Sabrina Huber from Department of Biological Engineering at Massachusetts Institute of Technology for critical editing of the manuscript. This work was supported by the National Natural Science Foundation of China (31300081, 31570074, 31600064), the National Program on Key Basic Research Project (973 programs, 2013CB734000), Open Project of State Key Laboratory of Microbial Metabolism from Shanghai Jiao Tong University (MMLKF13-05), the Initial Foundation of Doctoral Scientific Research in Anhui University (01001904, J01001935), and the National Innovation Experiment Program for University Students (J10118516053).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
conflict of interest
The authors declare that they have no competing interests.
Research involving human and animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, Y., Tan, G., Ke, M. et al. Enhanced lincomycin production by co-overexpression of metK1 and metK2 in Streptomyces lincolnensis. J Ind Microbiol Biotechnol 45, 345–355 (2018). https://doi.org/10.1007/s10295-018-2029-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2029-1