
Abstract
Complex reduced polyketides represent the largest class of natural products that have applications in medicine, agriculture, and animal health. This structurally diverse class of compounds shares a common methodology of biosynthesis employing modular enzyme systems called polyketide synthases (PKSs). The modules are composed of enzymatic domains that share sequence and functional similarity across all known PKSs. We have used the nomenclature of synthetic biology to classify the enzymatic domains and modules as parts and devices, respectively, and have generated detailed lists of both. In addition, we describe the chassis (hosts) that are used to assemble, express, and engineer the parts and devices to produce polyketides. We describe a recently developed software tool to design PKS system and provide an example of its use. Finally, we provide perspectives of what needs to be accomplished to fully realize the potential that synthetic biology approaches bring to this class of molecules.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Synthetic biology is an emerging discipline that combines the fields of biology, biotechnology and engineering for the design and construction of new biological parts, devices, and systems, as well as the re-design of existing natural biological systems, all for useful purposes. To make the engineering of biology faster, more reliable, and more reproducible, synthetic biologists have developed a framework for engineering biology that is similar to other engineering disciplines, including standardized, off the shelf parts (e.g., genes) with standardized connections that could be used and reused in the construction of devices and larger systems. For example, a computer can be assembled from standardized parts and devices (sound cards, processors, operating systems, hard drives, etc.) available from many vendors; devices such as monitors, scanners, printers, etc., are connected to the computer through a standard port.
Synthetic biology has numerous applications: it can be used for the construction of proteins that can serve as new vaccines, or new biochemical pathways synthesizing small molecules that can serve as pharmaceuticals, chemicals (solvents, adhesives, food flavorings, cosmetics, etc.), or biofuels, plant and animal growth stimulators or insecticides. Additional applications include the generation of new biosensors for early detection of medical conditions, or to detect environmental pollution. The hosts used for production of new proteins, small molecules, or biosensors were designated as chassis. Synthetic biology also includes the direct engineering of chassis, as well as the technologies such as CRISPR/Cas9 to make chassis easier to engineer.
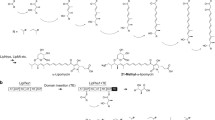
Reduced polyketides are a sub-class of natural products that have a variety of medical, veterinary, and agricultural applications. Examples, shown in Fig. 1, include the US Food and Drug Administration (FDA)-approved antibacterial agents erythromycin, fidaxomicin, rifamycin and tylosin; the antifungal agent amphotericin; the immunosuppressive agents rapamycin and tacrolimus; the cholesterol lowering agent lovastatin; the anthelmintic agent avermectin; the insecticide spinsosad; and monensin, a growth promotant for ruminants and coccidiostat in poultry. Curacin, discodermolide, epothilone, geldanamycin, and salinosporamide have anti-cancer properties and are in clinical trials. With the exception of lovastatin, which is produced by the fungus Aspergillus terreus, as well as a number of higher plants, all of the compounds shown in Fig. 1 are produced by bacteria, in particular members of the mycelia-forming family called actinomycetes that includes the most common genus Streptomyces, as well as the genera Dactylosporangium, and Saccharopolyspora, among others. Many members of the myxobacteria family also produce polyketides (e.g., epothilone, disorazole). Curacin is made in the cyanobacterium Lyngbya majuscula. Discodermolide was isolated from a sponge, but it is believed that it is produced by a symbiotic bacterium. Although these compounds do not resemble each other, and have different biological targets, they are all made by a class of enzymes known as modular polyketide synthases (PKSs), which produce the polyketide backbones. An example of a modular PKS is 6-deoxyerythronolide B synthase (DEBS), which produces 6-deoxyerythronolide B (6-dEB), the polyketide backbone of erythromycin (Fig. 2), which undergoes post-polyketide processing (glycosidation, hydroxylation, O-methylation) to generate the final product erythromycin. Some of the other compounds shown in Fig. 1 (e.g., chalcomycin, fidaxomicin, methymycin, and tylosin) undergo similar post-polyketide processing, whereas others, such as splenocin and geldanamycin undergo acylation or carbamoylation, respectively. Discodermolide and epothilone undergo little or no further elaboration. Some polyketides are connected to an amino acid or amino acid derivative (e.g., epothilone, rapamycin, tacrolimus, and lipomycin) via a linked non-ribosomal peptide synthetase (NRPS). The polyketide backbone of monensin reforms into a polyether, whereas part of the backbone of the reveromycin assumes a spiroketal configuration.

Structures of natural polyketides. The parts of the compounds determined by the corresponding polyketide synthase are highlighted in dark

Organization of the 6-deoxyerythronolide B synthase (DEBS) and structure of its product, 6-deoxyerythronolide B (6dEB). Stepwise growth of the polyketide chain of the polyketide chain is shown attached to the ACP domain of each module after full elaboration, including stereochemistry, by modular functions. Modules are labeled Load through Module 6 and are delineated by solid black lines. The proteins comprising DEBS are shown by arrows. The atoms labeled 1 and 13 in the acyl chain attached to module 6 undergo a lactonization reaction conducted by the TE domain to result in the production of the macrolactone 6-dEB, which undergoes further biochemical processing (not shown) to generate erythromycin (Fig. 1). Abbreviations: ACP, acyl carrier protein; AT, acyltransferase; DH, dehydratase; ER, enoyltransferase; KR, β-ketoreductase; KS, β-acyl ACP synthase; TE, thioesterase
Although great structural diversity is observed for polyketides, they share, as outlined below, an overarching similarity in their biosynthesis. This has drawn the attention of a variety of scientists interested in understanding the fundamental basis of how these molecules are made and the common and differentiating features of their biosynthesis. In addition, their medical and industrial importance has drawn the interest of pharmaceutical and industrial companies who desire to apply genetic approaches to change the structures of polyketides to improve their properties or generate chemical diversity.
This paper is intended for people interested in synthetic biology who may not be familiar with polyketides and PKSs. It is, therefore, not intended to be comprehensive with respect to the detailed enzymology of PKS or present a complete review of the genetic engineering of PKS genes that has been reported over the past 27 years. For that, the reader is directed to a number of recent reviews [3, 4, 21, 38, 44]. Rather, we attempt to show here that nature has devised a near perfect synthetic biology system of composable parts and devices in PKSs which can be assigned specific nomenclature according to function. We believe that this nomenclature will be interesting to synthetic biologists, as well as people who work with polyketides and PKSs, and useful for those interested in recomposing parts and devices to create new polyketides. We present an overview of work done in this area and discuss what needs to be understood that will make PKSs easier to engineer.
Overview of modular PKS enzymology
As can be seen in Fig. 2, 6-dEB is built by the stepwise condensation of propionyl-CoA and 6 molecules of methylmalonyl-CoA in a manner similar to the biosynthesis of fatty acids, which are produced by fatty acid synthases (FASs). However, unlike FASs [5], which use a single set of enzymes or enzymatic domains for all elongation steps, each polyketide chain elongation step is carried out by a discrete set of catalytic domains, which is defined as an extension module. Other modular PKSs also use acyl-CoAs or carboxyacyl-CoAs as substrates. The number of modules in a given PKS determines the size of the resultant polyketide produced, and the specific carboxyacyl-CoAs employed, which are selected by the modules themselves, determine the overall structure. DEBS has 6 extension modules that use methylmalonyl-CoA, in addition to the N-terminal loading module and the C-terminal off-loading domain, which accounts for the 14-membered macrolactone ring and a methyl group at every other position in the ring. In contrast, the fidaxomicin PKS, which, like erythromycin, also starts its synthesis with propionyl-CoA, has 8 extension modules, and uses malonyl-CoA in the first, sixth, and seventh extensions, methylmalonyl-CoA, in the second, third, fifth, and eighth extensions, and ethylmalonyl-CoA in the fourth extension, yielding an 18-membered macrolactone (Fig. 1). Each module also controls the reduction state of the corresponding elongated polyketide intermediate by having a specific set of reductase domains, whereas FASs exclusively provide only the fully reduced forms of their products. In addition, modular PKSs can control the stereochemistry at each chiral center as described below. Hence, the modular nature of the PKSs not only accounts for the thousands of diverse natural polyketide structures, but also offers a wealth of engineering opportunities for the production of novel compounds.
Polyketide chain elongation is catalyzed by three different catalytic domains: a ketosynthase (KS) domain, an acyltransferase (AT) domain, and an acyl carrier protein (ACP) domain where the AT domain determines the specific carboxyacyl-CoA incorporated into the growing polyketide chain, and then transfers it to the ACP in the same module. The KS domain then catalyzes decarboxylative C–C bond formation between the growing polyketide chain and the extender unit on the ACP. Modular PKSs are categorized into cis- and trans-AT (or AT-less) modular PKS systems. In the trans-AT type, each module lacks an AT domain and instead receives its substrate, by one or more free-standing ATs. For example, disorazole (Fig. 1) is produced by a trans-AT PKS. Although most of the free-standing ATs thus far identified are specific for malonyl-, methylmalonyl-, or ethylmalonyl-CoA [17], over 20 different malonyl-CoA analogs have been found to be incorporated into naturally occurring polyketides by cis-AT PKSs [46]. These analogs include short to medium-chain alkylmalonyl-CoAs, halogenated malonyl-CoAs, and benzylmalonyl-CoA (Fig. 3). Examples of compounds that contain side-chains in the polyketide backbone that are larger than a simple methyl group in Fig. 1 include tylosin and monensin (ethyl), rapamycin (methoxy), tacrolimus (methoxy and allyl), reveromycin (butyl, pentyl, isopentyl, or hexyl), splenomycin (benzyl), and salinosporamide (chloro-, fluoro-, or bromoethyl), all of which are introduced as malonyl-CoA or malonyl-ACP analogs by their corresponding cis-AT domains in their respective PKSs. In addition, there are a number of AT domains that can employ more than a single derivative of malonyl-CoA as substrate for incorporation, such as the AT domain of module 4 (AT4) of the epothilone PKS, which can use either malonyl- or methylmalonyl-CoA, the AT5 domain of the monensin PKS, which incorporates either methylmalonyl- or ethylmalonyl-CoA, and the AT5 domain of the reveromycin PKS which can utilize four different alkylated malonyl-CoAs as substrates. The DEBS AT domains are known to incorporate only the (2S)-enantiomer of methylmalonyl-CoA [27] and the subsequent KS-catalyzed condensation reactions generate (2R)-2-methyl-3-ketoacyl-ACP intermediates, the opposite stereochemical orientation [45]. It is also thought, but not yet proven, that all AT domains that use alkylated malonyl-CoAs as substrates only use the corresponding 2S-enantiomers.

Structures of malonyl-CoA analogs that are incorporated during biosynthesis of naturally occurring polyketides
After each condensation step, the newly generated β-keto group is reduced by a ketoreductase (KR) domain if present in the module. DEBS contains a KR domain in modules 1,2,4,5, and 6, and an inactive (reductase incompetent) KR domain in module 3 (Fig. 2). Importantly, the stereochemistries of both β-hydroxyl- and α-alkyl side-chains in the nascent polyketide generated are controlled primarily by the individual KR domains of each PKS module [20]. KR domains are characterized based on their stereochemical outcomes. A-type and B-type KRs generate the l-β-hydroxyl and d-β-hydroxyl groups, respectively. Each KR type can also control stereochemical orientations of α-substituents. This is important in all modules any acyl-CoA other than malonyl-CoA is introduced. A1- or B1-type KRs produce the d-α-substituent, and A2- or B2-type KRs produce l-α-substituent. Finally, C1- or C2-type KRs are reductase-incompetent but retain epimerase activity. The d/l system is not perfect but preferred over the R/S system because the assignment of R or S depends on the substituents present.
The trans or cis-double bonds within polyketides are generated by a dehydratase (DH) domain after KR-catalyzed reductions. DEBS has a DH domain in module 4. The syn-coplanar water elimination reaction from a substrate possessing d-β-hydroxyl group generates a trans-double bond [43]. Thus, a trans-double bond typically results from a module that contains B-type KR (B type KRs but not B2). In a module that contains an A-type KR, a cis-double bond can be generated. If a module further contains an enoyl reductase (ER) domain (e.g., DEBS module 4), stereoselective reduction of a trans-double bond possessing a α-substituent may occur [25]. ER domains are also characterized based on their stereochemical outcomes. L-type ERs generate l-α-substituents, and D-type ERs generate d-α-substituents. Recently, cryptic methyl group epimerase function of DH domains has also been reported [49].
Both cis- and trans-AT PKS modules are known to harbor an integrated, S-adenosylmethionine-dependent C-methyltransferase (MT) domain, which installs a methyl group to the α-carbon of β-keto polyketide intermediates. For example, module 8 of the epothilone PKS (EPOS M8) carries a C-MT domain. Since the AT domain specifies methylmalonyl-CoA, the gem-dimethyl group seen in epothilone is the result of a single methylation [37]. On the other hand, the C-MT domain of the PKS module for yersiniabactin biosynthesis specifies dimethylation to produce the corresponding gem-dimethyl group [37]. Recently, C-MTs from trans-AT PKS systems have been shown to be stereospecific [50].
After the above-mentioned reactions, the elongated and reduced (if reduction domains are present) polyketide intermediate is eventually translocated to the KS domain of the next module and similar reactions are repeated until the chain is fully elongated. Finally, thioesterase (TE) domains release products by cyclization or hydrolysis, thus generating lactones, lactams, and carboxylic acids [9]. TE domains are usually embedded into the last module of the PKSs but polyether-producing PKSs such as the nanchangmycin and monensin PKSs employ free-standing TEs to cleave the final intermediates [34, 42]. A few other less common termination mechanisms are also known. A tandem sulfotransferase (ST)-TE didomain yield terminal olefins, as found in the curacin PKS [7]. A reductive (R) domain produces aldehydes or alcohols, which are found in the coelimycin and myxalamid PKSs [14, 40].
The polyketide chain initiation event is catalyzed by domains, commonly called the loading module, preceding the first extension module. The most common loading module consists of a condensation-incompetent KS domain (e.g., KSQ, a Cys to Glu variant of a KS domain), an AT domain, and an ACP domain [31]. Here, the AT domain is specific for malonyl-CoA or methylmalonyl-CoA and generates acetyl or propionyl starter units, respectively, after a decarboxylation reaction by the KSQ domain. On the other hand, loading modules consisting of AT-ACP didomains tends to show broader substrate specificity (the AT lacks the conserved arginine residue, which stabilizes the acid moiety of malonyl- and methylmalonyl-CoA substrates). For example, as can been seen in the avermectin compounds shown in Fig. 1, the avermectin PKS can initiate polyketide biosynthesis from either isobutyryl-CoA or 2-methylbutyryl-CoA [18]. This PKS is also known to incorporate a huge variety of synthetic starters that can be exogenously fed to a variant of the producing organism that lacks the ability to make the natural starter units [10]. Similarly, the lipomycin PKS has also been demonstrated to have broad substrate specificity [52, 53]. On the other hand, the borrelidin PKS loading module was shown to be stereoselective and incorporate a carboxyacyl-CoA [15]. Another type of loading didomain is composed of a CoA-ligase (CoL) and an ACP domain, which usually activates a shikimic acid-derived cyclocarboxylic acid in an ATP-dependent manner and loads it onto the ACP. Some PKSs contain an additional domain between CoL and ACP domains, as found in the rapamycin PKS [39].
Synthetic biology of PKS: parts, devices, chassis
In synthetic biology jargon, the simplest unit of function is a part. For modular PKSs, the catalytic domains ACP, AT, DH, ER, KR, KS, C-MT, and TE, could be considered parts. A representative list of common PKS parts is shown in Table 1. AT parts are distinguishable on the basis of the substrate(s) that they utilize, e.g., malonyl-CoA, methylmalonyl-CoA, propionyl-CoA, etc. KR parts are distinguished on the basis of the stereochemistry generated at both the alkyl side chain (methyl, ethyl, etc.) at the α position, and the OH group at the β position. KR parts with stereochemistry unknown to the authors at the time of writing are represented by the code KRu. Other parts are represented in Table 1 by a single code including ER (the current version of our software cannot distinguish different ER types), but it is possible that different KS domains will be found to have different preferences for various acyl-ACPs. Similarly, subtypes of the other domains may also emerge.
A device consists of two or more parts, that has a function beyond that of the part. A simple device would be a gene, its promoter and a terminator, which can produce the corresponding protein. A more complicated device would be an operon consisting of several genes, along with a promoter and a terminator. In the context of PKS synthetic biology, PKS modules are considered devices. The simplest device in modular PKSs would consist of three parts: KS, AT and ACP; a complex device would contain KS, AT, DH, ER, KR, and ACP. A full PKS, which produces a functional polyketide and composed of an assemblage of devices would be considered a system. Without reference to PKS systems, the term module has also been used to describe a reusable device, built from simpler parts. In the discussion of PKSs, we will use the terms module and device interchangeably. Table 2 shows a list of 56 devices that are components of known PKSs, along with their cognate parts. Devices are grouped by presence of reductive domains with DH-ER-KR devices designated δ, DH-KR devices designated γ, KR devices designated β, and other devices designated α. The large number of devices is based on the variability of the AT and KR parts that comprise them. Figure 4 shows the structures of the growing or starting acyl chains attached to their cognate ACPs imparted by 25 devices selected from Table 2. It can be seen that devices α4 and α14 yield the structure, propionyl-ACP. In device α4, the ATp part (domain) directly loads propionyl-CoA to the ACP, whereas in device α14, the ATmm domain loads methylmalonyl-CoA onto the ACP and the KSQ domain subsequently decarboxylates it to leave propionyl-CoA.

Structures, including stereochemistry, of the α and β positions (shown for structure determined by device α8) of nascent polyketide chains determined by the devices shown (Table 2)
Table 3 lists the PKS devices used for the production of 15 of the polyketides shown in Fig. 1, excluding terminal domains. The smallest and largest PKSs shown encode the backbones for methymycin and amphotericin B and contain 6 and 19 devices, respectively. Many of the devices are common to two or more PKSs. Device γ3 is used in nine of the 12 PKSs shown, and is tandemly repeated in several PKSs including four times in lipomycin and six times in amphotericin. It is important to note that the PKS devices do not each have a single, unique sequence either at the protein or DNA level, although there is a high degree of similarity among the various representatives of each device. PKS devices are determined purely by function, namely the structure imparted onto the α and β carbons in the growing acyl chain during polyketide synthesis. The repetitive use of functionally identical modules to generate the large variety of polyketides produced in nature highlights them as natural reusable devices.
As mentioned above, many polyketides are generated in members of the genus Streptomyces. Streptomyces strains are likely the most promising chassis for forward engineering because they generally encode a substrate promiscuous phosphopantetheinyl transferase (PPTase) that is necessary to functionalize heterologous PKS genes expressed in the host. PPTases are absent in the model microbial hosts Escherichia coli and Saccharomyces cerevisiae widely used in synthetic biology. Streptomyces hosts are also known to produce diverse acyl-CoAs and carboxyacyl-CoAs such as isobutyryl-CoA and methylmalonyl-CoA that are not naturally produced in E. coli and S. cerevisiae. Recently, approximately 200 native or synthetic promoters and ~ 200 ribosomal binding sites were characterized in Streptomyces venezuelae, and some were also validated in two other Streptomyces strains; their promoter strengths were highly correlated in the three chassis [1]. CRISPR/Cas9 systems have been developed for genome editing of several different Streptomyces strains [26]. These systems enabled direct selection (via survival) for double homologous recombination, and were shown to shorten the time and effort required for genome engineering over the methodologies formerly used for gene replacement. For large gene insertions (> 5 kb), a number of actinophage integrase-based site-specific gene integration systems have been developed and used for heterologous polyketide production in Streptomyces [2, 36]. Many replicating plasmids, as well as a temperature-sensitive plasmid have been developed into vectors, and methods to move DNA from E. coli to a variety of Streptomyces strains have also been developed [23].
Engineering PKSs
Synthetic biologists use the term refactoring to describe the engineering of genes or operons. The modularity of PKSs and the similarities of the devices that compose them render them as excellent targets for refactoring (at the DNA level) to produce novel compounds with predicted structures. Over the past 27 years, much work in this area has been undertaken, mainly employing cis-AT PKS systems. Most efforts have been directed at changing the parts within modules (AT or reduction domain swapping), deleting modules or combining heterologous PKS modules to form novel polyketides. It should be noted that, beyond the development of the technology, or the seeking of greater understanding of a specific PKS-mediated biochemical step, most of the reported efforts have been directed at producing novel antibiotics or other polyketides of medical interest. In these cases, it generally would not have been possible to predict a priori which novel molecules would have the desired properties, hence a combinatorial approach, where many compounds could be generated simultaneously, would be preferred. Because of simple, as well as sophisticated assays for activities that have been developed for polyketides of interest to various pharmaceutical companies, the emphasis has been on generating large numbers of novel compounds of diverse structure, where low-level production of is usually sufficient for assessment of potential value. Examples include the production of derivatives of erythromycin, rapamycin, and geldanamycin, among others [28, 35, 47]. Unfortunately, the molecular bases of why some engineered PKSs worked better than others in these in vivo experiments were generally not explored.
A less common approach to refactoring modular PKSs, and one in which our laboratory is heavily engaged, is to produce biofuels and other industrially important chemicals as drop-in replacements for currently used petroleum-based compounds. In these cases, only a single, or small number of novel compounds are targeted for production. Examples include adipic acid [16] and short-chain ketones such as MEK (2-butanone), and MIBK (4-methyl-2-pentanone) [51], which are used as solvents. In general, the monetary values of the molecules targeted are low, hence to be competitive with the same compounds resourced from petroleum, considerations of product titers, rates of production, and yields (relative molar amounts of product to carbon used in fermentation) are very important. Here, the emphasis is on production levels. Success heavily depends upon precise PKS engineering, employing a chassis capable of relatively fast growth rates and reaching high densities on inexpensive media, as well as possessing all the biochemical precursor pathways to support functional expression of heterologous PKS systems.
ClusterCAD
We have developed the online database ClusterCAD which facilitates the selection of natural cis-AT PKS parts to design a novel chimeric PKS that produces a desired small molecule [11]. ClusterCAD will identify a natural truncated PKS with a chemical intermediate most similar to a desired target, and can then identify potential donor parts for subsequent domain swaps, which are necessary to produce a desired target compound. Currently, ClusterCAD currently contains 72 PKS clusters. Notably, among those absent are the tacrolimus and salinosporamide PKS clusters.
Here we demonstrate the utility of ClusterCAD to guide the selection of PKS parts to produce the novel polyketide target shown in Fig. 5a. First, we use the ClusterCAD chemical structure search tool to identify a natural PKS as a starting point for engineering. One of the top hits, or most chemically similar natural PKSs in ClusterCAD to this target molecule is narbonolide, the pikromycin precursor produced by the PKS. The ClusterCAD chemical structure search tool produces a visual MCS plot (Maximum Common Substructure) which highlights the shared substructure between the query and each hit, as shown in Fig. 5b. As this plot visually highlights the similar regions between both structures, it is straightforward to identify the following sequence of engineering modifications to the pikromycin PKS, which would result in the desired product:

a Example chemical target used to demonstrate guiding chimeric PKS engineering with the ClusterCAD software tool. b A maximum common substructure (MCS) plot highlighting the common regions between the example chemical target (on right), and narbonolide, a chemically similar natural PKS found in ClusterCAD (on left). The non-highlighted regions represent engineering modifications necessary to produce the example product
(1) swap the module 0 (load module) acyltransferase (AT) domain for an AT which naturally incorporates an isobutyryl-CoA substrate; (2) swap the module 1 acyltransferase (AT) domain for an AT which naturally incorporates an ethylmalonyl-CoA substrate; (3) disable the module 5 ketoreductase (KR); (4) insert a type A1 ketoreductase (KR) in module 6; (5) swap the module 6 acyltransferase (AT) domain for an AT which naturally incorporates an allylmalonyl-CoA substrate.
After decomposing the difference between the natural PKS and the target compound into a sequence of engineering modifications as shown above, the ClusterCAD sequence search tool can identify suggested donor parts for each domain swap, based on amino acid sequence similarity. For domain swaps, this is done by searching the sequence of each domain for the most similar domain with a desired annotation, whereas for domain insertions you can search the entire ‘acceptor’ polypeptide for the most sequence similar polypeptide which includes the desired new domain annotations. This produces the following list of top match donor parts for the above modifications, in order: (1) Divergolide module 6 acyltransferase (AT); (2) Angolamycin module 5 acyltransferase (AT); (3) Tirandamycin module 1 ketoreductase (KR); (4) pikromycin module 5 ketoreductase (KR); (5) None, ClusterCAD does not yet contain any PKS clusters which incorporate allylmalonyl-CoA (or ACP). The AT domain of module 4 of the tacrolimus PKS specifies allylmalonyl-CoA or allylmalonyl-ACP [19, 32].
In summary, this example demonstrates the overall process of using ClusterCAD to design a chimeric PKS for a novel chemical target, by first identifying a chemically similar natural product, and then identifying a set of engineering modifications and donor parts which could, in principle yield the desired product. In practice, the rules which govern domain swap compatibility, and chimeric junction loci remain poorly understood, so it is necessary to test a large number of donor parts and junctions to yield an active enzyme which produces the desired product at detectable levels.
Conclusions
Theoretically, it should be possible to design a polyketide of desired structure, write out the list of parts and devices required from Tables 1 and 2, respectively (to resemble the strategies shown in Table 3 for known PKS systems), employ ClusterCAD or similar software to design the devices, synthesize the DNA of the required sequence, introduce the DNA into a desired chassis, and express it under a selected means of regulation. Except for the production of small molecules that are generated from three or fewer modules, or the limited engineering of one or two modules within the framework of a large PKS, this has not yet been reported. Whereas significant progress in our ability to productively alter the substrate specificities and reductive outcomes generated from within a module has taken place over the last few years [4], the ability to link modules in sequence to generate completely new polyketide structures without losing biochemical activity is still extremely challenging.
The first issue is the strategy to be employed to covalently link the modules so that there is efficient chain transfer. Small polyketides can be made by covalently assembling two PKS modules into a single polypeptide. It may be possible to assemble a larger set of modules into a single polypeptide (e.g., nysC has 6 modules in a single polypeptide [6]). This approach would obviate the need for docking domains to enable correct module–module interactions and a few successful examples have been reported in two module systems [13, 48]. Since the polypeptide chains of modular PKS systems assemble into homodimers [41], it is likely that the modules assembled in the long polypeptide will require placement of dimerization elements in appropriate positions if they are not already present [12, 54]. An empirical approach appears to be the only way to develop this strategy and should be undertaken.
Second, if construction of new PKSs requires the assembly of modules into three or more polypeptides, non-covalent intermodular interactions through docking domains may be necessary. However, previous efforts showed that more than 90% of the hybrid PKSs connected by docking domains showed < 10% activities compared to the wild-type counterparts in this approach [24, 29, 30]. It is likely that success here will require greater knowledge of the detailed structure of not only the docking domains but also of the modules themselves, and perhaps entire PKS systems. Controlling non-covalent intermodular interactions gives the greatest opportunity for rapidly generating large numbers of novel polyketides, and remains the only means for accomplishing this goal combinatorially, which would be a major achievement in the application of synthetic biology.
Finally, several other issues also need to be addressed to generate a working strategy for de novo production of novel polyketides. The first is chain passage of the growing polyketide chain from the ACP of one module to the correct KS domain immediately downstream. The accepting KS must make proper contact with the donating upstream ACP, and must be able to hold the incoming substrate in its active site to allow thiotransfer to its active site cysteine. The KS must catalyze chain elongation reactions at rates faster than its overall kinetics (or lose biochemical activity). Many KS domains are known to be somewhat substrate selective for both chain length and stereochemistry. It may be possible to replace substrate-selective KS domains in each module with those that are more substrate flexible (e.g., mycolactone KSs [33]), but that means non-cognate intramodular KS–ACP interactions for the required chain extensions. Because decreased kcat/Km values were previously observed in chain elongation with heterologous KS–ACP interfaces [8] and chain extension reaction is most likely the rate-limiting step in DEBS [22], it is very challenging to solve this issue.
References
Bai C, Zhang Y, Zhao X, Hu Y, Xiang S, Miao J, Lou C, Zhang L (2015) Exploiting a precise design of universal synthetic modular regulatory elements to unlock the microbial natural products in Streptomyces. Proc Natl Acad Sci USA 112:12181–12186. https://doi.org/10.1073/pnas.1511027112
Baltz RH (2010) Streptomyces and Saccharopolyspora hosts for heterologous expression of secondary metabolite gene clusters. J Ind Microbiol Biotechnol 37:759–772. https://doi.org/10.1007/s10295-010-0730-9
Barajas JF, Blake-Hedges JM, Bailey CB, Curran S, Keasling JD (2017) Engineered polyketides: synergy between protein and host level engineering. Synth Syst Biotechnol 2:147–166. https://doi.org/10.1016/j.synbio.2017.08.005
Bayly CL, Yadav VG (2017) Towards precision engineering of canonical polyketide synthase domains: recent advances and future prospects. Molecules. https://doi.org/10.3390/molecules22020235
Beld J, Lee DJ, Burkart MD (2015) Fatty acid biosynthesis revisited: structure elucidation and metabolic engineering. Mol BioSyst 11:38–59. https://doi.org/10.1039/c4mb00443d
Brautaset T, Sekurova ON, Sletta H, Ellingsen TE, StrLm AR, Valla S, Zotchev SB (2000) Biosynthesis of the polyene antifungal antibiotic nystatin in Streptomyces noursei ATCC 11455: analysis of the gene cluster and deduction of the biosynthetic pathway. Chem Biol 7:395–403
Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH (2004) Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod 67:1356–1367. https://doi.org/10.1021/np0499261
Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C (2006) Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J Am Chem Soc 128:3067–3074. https://doi.org/10.1021/ja058093d
Du L, Lou L (2010) PKS and NRPS release mechanisms. Nat Prod Rep 27:255–278. https://doi.org/10.1039/b912037h
Dutton CJ, Gibson SP, Goudie AC, Holdom KS, Pacey MS, Ruddock JC, Bu’Lock JD, Richards MK (1991) Novel avermectins produced by mutational biosynthesis. J Antibiot (Tokyo) 44:357–365
Eng CH, Backman TWH, Bailey CB, Magnan C, Garcia Martin H, Katz L, Baldi P, Keasling JD (2017) ClusterCAD: a computational platform for type I modular polyketide synthase design. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx893
Eng CH, Yuzawa S, Wang G, Baidoo EE, Katz L, Keasling JD (2016) Alteration of polyketide stereochemistry from anti to syn by a ketoreductase domain exchange in a Type I modular polyketide synthase subunit. Biochemistry 55:1677–1680. https://doi.org/10.1021/acs.biochem.6b00129
Gokhale RS (1999) Dissecting and exploiting intermodular communication in polyketide synthases. Science 284:482–485. https://doi.org/10.1126/science.284.5413.482
Gomez-Escribano JP, Song L, Fox DJ, Yeo V, Bibb MJ, Challis GL (2012) Structure and biosynthesis of the unusual polyketide alkaloid coelimycin P1, a metabolic product of the cpk gene cluster of Streptomyces coelicolor M145. Chem Sci 3:2716. https://doi.org/10.1039/c2sc20410j
Hagen A, Poust S, de Rond T, Yuzawa S, Katz L, Adams PD, Petzold CJ, Keasling JD (2014) In vitro analysis of carboxyacyl substrate tolerance in the loading and first extension modules of borrelidin polyketide synthase. Biochemistry 53:5975–5977. https://doi.org/10.1021/bi500951c
Hagen A, Poust S, Rond T, Fortman JL, Katz L, Petzold CJ, Keasling JD (2016) Engineering a polyketide synthase for in vitro production of adipic acid. ACS Synth Biol 5:21–27. https://doi.org/10.1021/acssynbio.5b00153
Helfrich EJ, Piel J (2016) Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep 33:231–316. https://doi.org/10.1039/c5np00125k
Ikeda H, Nonomiya T, Usami M, Ohta T, Omura S (1999) Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis. Proc Natl Acad Sci USA 96:9509–9514
Jiang H, Wang YY, Guo YY, Shen JJ, Zhang XS, Luo HD, Ren NN, Jiang XH, Li YQ (2015) An acyltransferase domain of FK506 polyketide synthase recognizing both an acyl carrier protein and coenzyme A as acyl donors to transfer allylmalonyl and ethylmalonyl units. FEBS J 282:2527–2539. https://doi.org/10.1111/febs.13296
Keatinge-Clay AT (2016) Stereocontrol within polyketide assembly lines. Nat Prod Rep 33:141–149. https://doi.org/10.1039/c5np00092k
Keatinge-Clay AT (2017) The uncommon enzymology of cis-acyltransferase assembly lines. Chem Rev 117:5334–5366. https://doi.org/10.1021/acs.chemrev.6b00683
Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE (2007) Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu Rev Biochem 76:195–221. https://doi.org/10.1146/annurev.biochem.76.053105.093515
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich, UK
Klaus M, Ostrowski MP, Austerjost J, Robbins T, Lowry B, Cane DE, Khosla C (2016) Protein–protein interactions, not substrate recognition, dominate the turnover of chimeric assembly line polyketide synthases. J Biol Chem 291:16404–16415. https://doi.org/10.1074/jbc.M116.730531
Kwan DH, Leadlay PF (2010) Mutagenesis of a modular polyketide synthase enoylreductase domain reveals insights into catalysis and stereospecificity. ACS Chem Biol 5:829–838. https://doi.org/10.1021/cb100175a
Liu Z, Liang Y, Ang EL, Zhao H (2017) A new era of genome integration-simply cut and paste! ACS Synth Biol 6:601–609. https://doi.org/10.1021/acssynbio.6b00331
Marsden AFA, Caffrey P, Aparicio JF, Loughran MS, Staunton J, Leadlay PF (1994) Stereospecific acyl transfers on the erythromycin-producing polyketide synthase. Science 263:378–380. https://doi.org/10.1126/science.8278811
McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Betlach M, Ashley G (1999) Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel ‘‘unnatural’’ natural products. Proc Natl Acad Sci USA 96:1846–1851
Menzella HG, Carney JR, Santi DV (2007) Rational design and assembly of synthetic trimodular polyketide synthases. Chem Biol 14:143–151. https://doi.org/10.1016/j.chembiol.2006.12.002
Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA, Santi DV (2005) Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotechnol 23:1171–1176. https://doi.org/10.1038/nbt1128
Moore BS, Hertweck C (2002) Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat Prod Rep 19:70–99. https://doi.org/10.1039/b003939j
Motamedi H, Shafiee A (1998) The biosynthetic gene cluster for the macrolactone ring of the immunosuppressant FK506. Eur J Biochem 256:528–534
Murphy AC, Hong H, Vance S, Broadhurst RW, Leadlay PF (2016) Broadening substrate specificity of a chain-extending ketosynthase through a single active-site mutation. Chem Commun 52:8373–8376. https://doi.org/10.1039/c6cc03501a
Oliynyk M, Stark CBW, Bhatt A, Jones MA, Hughes-Thomas ZA, Wilkinson C, Oliynyk Z, Demydchuk Y, Staunton J, Leadlay PF (2003) Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin in Streptomyces cinnamonensis and evidence for the role of monB and monC genes in oxidative cyclization. Mol Microbiol 49:1179–1190. https://doi.org/10.1046/j.1365-2958.2003.03571.x
Patel K, Piagentini M, Rascher A, Tian ZQ, Buchanan GO, Regentin R, Hu Z, Hutchinson CR, McDaniel R (2004) Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem Biol 11:1625–1633. https://doi.org/10.1016/j.chembiol.2004.09.012
Phelan RM, Sachs D, Petkiewicz SJ, Barajas JF, Blake-Hedges JM, Thompson MG, Reider Apel A, Rasor BJ, Katz L, Keasling JD (2017) Development of next generation synthetic biology tools for use in Streptomyces venezuelae. ACS Synth Biol 6:159–166. https://doi.org/10.1021/acssynbio.6b00202
Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, Keasling JD (2015) Divergent mechanistic routes for the formation of gem-dimethyl groups in the biosynthesis of complex polyketides. Angew Chem Int Ed Engl 54:2370–2373. https://doi.org/10.1002/anie.201410124
Robbins T, Liu YC, Cane DE, Khosla C (2016) Structure and mechanism of assembly line polyketide synthases. Curr Opin Struct Biol 41:10–18. https://doi.org/10.1016/j.sbi.2016.05.009
Schwecke T, Aparicio JF, Molnar I, Konig A, Khaw LE, Haydock SF, Oliynyk M, Caffrey P, Cortes J, Lester JB et al (1995) The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proc Natl Acad Sci USA 92:7839–7843
Silakowski B, Nordsiek G, Kunze B, Blocker H, Muller R (2001) Novel features in a combined polyketide synthase/non-ribosomal peptide synthetase: the myxalamid biosynthetic gene cluster of the myxobacterium Stigmatella aurantiaca Sga15. Chem Biol 8:59–69. https://doi.org/10.1016/S1074-5521(00)00056-9
Staunton J, Caffrey P, Aparicio JF, Roberts GA, Bethell SS, Leadlay PF (1996) Evidence for a double-helical structure for modular polyketide synthases. Nat Struct Biol 3:188–192
Sun Y, Zhou X, Dong H, Tu G, Wang M, Wang B, Deng Z (2003) A complete gene cluster from Streptomyces nanchangensis NS3226 encoding biosynthesis of the polyether ionophore nanchangmycin. Chem Biol 10:431–441. https://doi.org/10.1016/s1074-5521(03)00092-9
Valenzano CR, You YO, Garg A, Keatinge-Clay A, Khosla C, Cane DE (2010) Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. J Am Chem Soc 132:14697–14699. https://doi.org/10.1021/ja107344h
Weissman KJ (2016) Genetic engineering of modular PKSs: from combinatorial biosynthesis to synthetic biology. Nat Prod Rep 33:203–230. https://doi.org/10.1039/c5np00109a
Weissman KJ, Timoney M, Bycroft M, Grice P, Hanefeld U, Staunton J, Leadlay PF (1997) The molecular basis of Celmer’s rules: the stereochemistry of the condensation step in chain extension on the erythromycin polyketide synthase. Biochemistry 36:13849–13855. https://doi.org/10.1021/bi971566b
Wilson MC, Moore BS (2012) Beyond ethylmalonyl-CoA: the functional role of crotonyl-CoA carboxylase/reductase homologs in expanding polyketide diversity. Nat Prod Rep 29:72–86. https://doi.org/10.1039/c1np00082a
Wlodek A, Kendrew SG, Coates NJ, Hold A, Pogwizd J, Rudder S, Sheehan LS, Higginbotham SJ, Stanley-Smith AE, Warneck T, Nur EAM, Radzom M, Martin CJ, Overvoorde L, Samborskyy M, Alt S, Heine D, Carter GT, Graziani EI, Koehn FE, McDonald L, Alanine A, Rodriguez Sarmiento RM, Chao SK, Ratni H, Steward L, Norville IH, Sarkar-Tyson M, Moss SJ, Leadlay PF, Wilkinson B, Gregory MA (2017) Diversity oriented biosynthesis via accelerated evolution of modular gene clusters. Nat Commun 8:1206. https://doi.org/10.1038/s41467-017-01344-3
Wu N, Tsuji SY, Cane DE, Khosla C (2001) Assessing the balance between protein–protein interactions and enzyme-substrate interactions in the channeling of intermediates between polyketide synthase modules. J Am Chem Soc 123:6465–6474
Xie X, Garg A, Khosla C, Cane DE (2017) Elucidation of the cryptic methyl group epimerase activity of dehydratase domains from modular polyketide synthases using a tandem modules epimerase assay. J Am Chem Soc 139:9507–9510. https://doi.org/10.1021/jacs.7b05502
Xie X, Khosla C, Cane DE (2017) Elucidation of the stereospecificity of C-methyltransferases from trans-AT polyketide synthases. J Am Chem Soc 139:6102–6105. https://doi.org/10.1021/jacs.7b02911
Yuzawa S, Deng K, Wang G, Baidoo EE, Northen TR, Adams PD, Katz L, Keasling JD (2017) Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth Biol 6:139–147. https://doi.org/10.1021/acssynbio.6b00176
Yuzawa S, Eng CH, Katz L, Keasling JD (2013) Broad substrate specificity of the loading didomain of the lipomycin polyketide synthase. Biochemistry 52:3791–3793. https://doi.org/10.1021/bi400520t
Yuzawa S, Eng CH, Katz L, Keasling JD (2014) Enzyme analysis of the polyketide synthase leads to the discovery of a novel analog of the antibiotic alpha-lipomycin. J Antibiot (Tokyo) 67:199–201. https://doi.org/10.1038/ja.2013.110
Zheng J, Fage CD, Demeler B, Hoffman DW, Keatinge-Clay AT (2013) The missing linker: a dimerization motif located within polyketide synthase modules. ACS Chem Biol 8:1263–1270. https://doi.org/10.1021/cb400047s
Acknowledgements
This work was supported by the Joint BioEnergy Institute, which is funded by the US Department of Energy (DOE), the Office of Science, Office of Biological and Environmental Research under Contract No. DE-AC02-05CH11231 between DOE and Lawrence Berkeley National Laboratory. The publisher, by accepting the article for publication, acknowledges that the US government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of the manuscript, or allow others to do so, for US government purpose. The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yuzawa, S., Backman, T.W.H., Keasling, J.D. et al. Synthetic biology of polyketide synthases. J Ind Microbiol Biotechnol 45, 621–633 (2018). https://doi.org/10.1007/s10295-018-2021-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2021-9