
Abstract
A combined system of a unique dye-decolorizing peroxidase (Ftr-DyP) and a laccase obtained from the basidiomycete Funalia trogii converted the precursor (+)-valencene completely to the high-value grapefruit flavour constituent (+)-nootkatone, reaching a concentration maximum of 1100 mg/L. In the presence of 1 mM Mn2+ and 2.5 mM p-coumaric acid, (+)-nootkatone was the predominating volatile product, and only traces of substrate and the nootkatols were detectable after 24 h. Hence, the two-enzyme-system reproduced the oxidizing activity observed before for the crude culture supernatant. The newly discovered Ftr-DyP was purified, sequenced and further characterized as a thermostable, non-glycosylated protein with a pH-optimum in the acidic range and a calculated mass of 52.3 kDa. Besides the typical activity of DyPs towards anthraquinone dyes, Ftr-DyP also oxidized Mn2+ and showed activity in the absence of hydrogen peroxide. Neither the DyP from Mycetinis scorodonius nor the manganese peroxidase from Nematoloma frowardii were able to replace Ftr-DyP in this reaction. A hypothetical reaction mechanism is presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The allylic oxidation of (+)-valencene to produce the grapefruit-like character-impact aroma compound (+)-nootkatone (Fig. 1) without using hazardous oxidants, such as dichromate, has been a kind of holy grail of flavour biotechnology for many years [15]. Biocatalysis not only is more environmentally friendly and sustainable, but also complies with the legal requirements in Europe and the US for the attribute “natural” of a flavour compound. In view of the strong industrial interest in natural nootkatone, many bacterial, yeast or plant cells were reported to perform the aspired reaction, more recently including recombinant and engineered cells [34]. These systems suffer from severe disadvantages, such as slow growth of the biocatalyst, volatility of substrate and product in aerated culture, cytotoxicity of substrate and product, product degradation after formation, multiple metabolic pathways resulting in mixed product formation, and ultimately low productivities. Attempts to circumvent these problems were based on isolated enzymes [16]. The major hurdle in an enzyme based oxidation is the requirement for a cofactor and its regeneration. An early example was a laccase mediator system which was patented and yielded 28.6% (w/w) using the explosive hydroxybenzotriazole as a synthetic mediator [9]. An advanced one-pot two enzyme system used a cytochrome P450 monooxygenase for the hydroxylation step to nootkatol, followed by an alcohol dehydrogenase step with cofactor regeneration. A (+)-nootkatone concentration of 360 mg/L was reached [28]. Another approach used lyophilisate of the fungus Pleurotus sapidus, which contained a lipoxygenase catalysing the insertion of oxygen followed by a Schenck rearrangement and hydroperoxide decay [6]. Yields of up to 320 mg/l after an incubation time of 24 h [6] or 28 mol-% after 48 h, respectively, were achieved [6, 21].

Oxidation of (+)-valencene (1) resulting in the formation of (+)-nootkatone (2) and nootkatols (3)
In the present work a well-defined system consisting of a dye-decolorizing peroxidase (Ftr-DyP) and a laccase (Ftr-Lcc) from the fungus Funalia trogii is described which converted (+)-valencene efficiently to (+)-nootkatone with a high selectivity and with very little formation of volatile side-products. While the process-stable and high-redox laccase from Funalia was previously characterized and applied to the crosslinking of proteins [30] and the oxidative cleavage of curcumin to vanillin [3], nothing was known about the Ftr-DyP. Thus, it was the aim to heterologously express and to characterize this unique enzyme to prepare for a possible future industrial application.
Materials and methods
Chemicals
If not stated otherwise, chemicals were obtained either from Sigma-Aldrich, Roth or Fluka in p.a. quality.
Organism and cultivation
The Funalia trogii strain (no. 11919) was obtained from the German Collection of Microorganisms and Cell Cultures (DMSZ). The fungus was pre-grown on an agar plate containing standard nutrient liquid (SNL) medium [29]. A submerged pre-cultivation flask was prepared by transferring 1 cm2 of the mycelium grown agar to 125 mL of liquid SNL medium and homogenised by ULTRA-TURRAX stirring. After 7 days of cultivation approximately 10 mL of pre-cultivated medium was transferred into 1 L Erlenmeyer flasks containing 500 mL SNL medium with additional 30 g/L wheat bran and 0.1 mM CuSO4 to enhance Ftr-Lcc and Ftr-DyP production based on previous evidence [20, 30]. Cultivations were carried out at 24 °C at 150 rpm.
Isolation and purification of the Ftr-DyP
The culture supernatant was harvested at maximal Ftr-DyP activity (22 days) and separated from the fungal mycelium through centrifugation at 5000×g. It was then mixed with an ammonium sulfate solution to a concentration of 280 g/L. After 4 h of incubation at 4 °C the precipitated protein was isolated by centrifugation at 5000×g and re-dissolved in 20 mM TRIS buffer of pH 8.0. Precipitation of the dissolved protein was repeated with 250 g/L ammonium sulfate.
The dissolved precipitate was dialysed overnight (VISKING® dialysis tubing 20/32, 12–14 kDa MWCO, SERVA) against 20 mM TRIS buffer of pH 8.0 and further purified by means of fast protein liquid chromatography (NGCTM, Biorad). Two millilitre of the sample were applied onto a HiTrap Q XL column of 1 mL (GE Healthcare) with a flowrate of 0.7 mL/min and 20 mM TRIS buffer of pH 8.0 as starting buffer. Stepwise elution was carried out with additional 1 M sodium chloride. Ftr-DyP containing fractions were identified by the manganese malonate assay, pooled and concentrated by ultrafiltration using a tangential flow membrane filter device with a 10 kDa cut-off (Amicon® Ultra-15, Merck Millipore). Further purification was attained by use of a Superdex 75 Increase 10/300 GL column (GE Healthcare). Samples of 250 µL were injected, and the runs were performed with 0.2 M acetate buffer of pH 5.0 at a flow rate of 0.5 mL/min.
Isolation and purification of the Ftr-Lcc
The culture supernatant (22 days of cultivation) was mixed with an ammonium sulfate solution to a concentration of 280 g/L and incubated for 4 h at 4 °C. Precipitated protein was removed through centrifugation at 5000×g. The remaining solution was filtered through a syringe filter (0.45 µm, cellulose acetate, VWR) and applied onto a 1 mL Phenyl FF column (GE Healthcare) using FPLC (NGCTM, Biorad). Runs were performed at a flowrate of 1 mL/min with 50 mM phosphate buffer of pH 6.5 containing 2 M ammonium sulfate as starting conditions, while elution was carried out with omission of ammonium sulfate. Laccase containing fractions were identified by the ABTS assay, collected and re-buffered by ultrafiltration to 20 mM BIS–TRIS buffer of pH 7.0. The sample was then further purified with a 1 mL HiTrap Q XL column with stepwise elution of additional sodium chloride up to a concentration of 1 M.
Gel electrophoresis and staining
Prior to gel electrophoretic analysis, samples were treated with endoglycosidase H (Endo H). One microlitre of an Endo H preparation was added to 20 µL of the sample and incubated for 2 h at 37 °C. After incubation protein samples were mixed in a ratio of 1:1 with a loading buffer containing 150 mM TRIS–HCl (pH 6.8), 200 mM DTT, 20% (v/v) glycerol, 4% (w/v) SDS and 25 mg/L bromophenol blue and heated for 10 min at 95 °C.
A 12% cross-linked discontinuous SDS gel was prepared according to Laemmli [14]. 20 µL of each sample were applied on the gel. Gel electrophoresis was performed at 20 mA (Mini-PROTEAN tetra cell, Bio-Rad) with a running buffer containing 25 mM TRIS, 1.9 M glycine and 1 g/L SDS. After the run, the gels were stained with a Coomassie Brilliant Blue solution (InstantBlueTM, Expedion). Protein masses were calculated through linear regression of protein markers in the range from 10 to 250 kDa (unstained Precision Plus ProteinTM Standards, Biorad).
Peptide mass fingerprinting
Peptide mass fingerprinting of the Ftr-DyP was performed using electrospray ionization-tandem mass spectrometry (ESI–MS/MS) with a quadrupole time of flight (QTOF) mass spectrometer (maXis impact, Bruker). The respective Ftr-DyP band was cut out from an SDS gel, washed with 30% (v/v) ethanol at 70 °C for 20 min and tryptically hydrolysed prior ESI–MS/MS analysis as reported previously [19].
Amplification of the Ftr-DyP
Extraction of total RNA from the mycelium of F. trogii from culture day 20 as well as cDNA synthesis were performed using the 3′-adapter primer for reverse transcription, unless stated otherwise, as previously described [1]. The peptide hits from ESI–MS/MS analysis were used for a blast search to identify similar sequences. Based on these sequences degenerated primer were designed (FtrDyP_for1 and FtrDyP_rev1) and used for the initial amplification of the gene. The PCR as well as the ligation conditions for subcloning were as described previously [1]. Primer sequences are shown in Table 1. The 3′ end of the gene was identified using the primer FtrDyP_for2 and 3′ anchor primer. The 5′ RACE was performed using a RNA-ligase mediated ligation of an 5′ adapter primer to the cDNA using a modified method based on Zhang and Chiang and Trout et al. [31, 35]. As a first step, 3 µg of the total RNA was transcribed using FtrDyP_rev2 as a specific primer, and afterwards the remaining RNA was degraded by adding 1 M NaOH and boiling for 5 min at 95 °C. After neutralizing the solution using 1 M HCl, the cDNA was purified using the innuPREP DOUBLEpure Kit (Analytik Jena). The 5′ adapter primer was phosphorylated using T4 polynucleotide phosphatase (Fischer Scientific), and the ligation was performed using 2 pmol phosphorylated primer, 0.1 mg/mL BSA, 1 U/µL T4 RNA Ligase, 0.8 ng/µL cDNA in RNA ligase buffer and PEG 6000 addition for 18 h at 18 °C.
The following amplification using the ligated primer-cDNA-complex as template was performed with the primer Adp nested for and FtrDyP_rev2 using a touchdown PCR (72–60 °C, 45 s elongation). Afterwards the PCR solution was diluted 1:10, and a nested PCR was performed using the primer Adp nested for and FtrDyP _rev3. The complete sequence was amplified from cDNA as well as gDNA using the primer FtrDyP_for3 and FtrDyP_rev4 cloned into pUC57 for long term storage and uploaded to GenBank (MF953832).
For the heterologous expression of the protein its coding gene was amplified using the primer FtrDyP_for_tata and FtrDyP_rev_8xHis_NotI and ligated into the expression vector pPIC9. While the vector was digested using SnaBI and NotI (ThermoFischer), the PCR amplificate was only digested using NotI before ligation. After the ligation using T4 DNA ligase, the vector was transformed into E. coli TOP10 cells for vector propagation [24], isolated (NucleoSpin, Macherey–Nagel, Düren, Germany) and subsequently transformed into P. pastoris GS115 cells (Invitrogen) using a standard protocol [17].
Expression analysis
To screen for the best producing clone, 96 transformants were examined in deep well plates for their expression rate. Each transformant was inoculated in 600 µL YEPD medium (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) dextrose) and grown to high density, typically ranging from OD600 1.5–2.0 (72 h, 28 °C, 300 rpm). Afterwards the medium was changed to BMMY (1.34% (w/v) yeast nitrogen base, 1% (w/v) yeast extract, 2% (w/v) peptone, 100 mM potassium phosphate, pH 6.0, 4 × 10−5 % (w/v) biotin, 1% (v/v) methanol) by resuspension of the cells (2000×g, 10 min) and induced with a daily addition of 1% (v/v) methanol for 120 h at 20 °C. The Ftr-DyP activity was measured daily with ABTS and hydrogen peroxide as substrates (cf. Enzyme activities).
Purification of the recombinant Ftr-DyP
Supernatant of P. pastoris culture was separated from the cells through centrifugation at 5000×g for 15 min. Binding buffer was added to contain a final concentration of 0.5 M NaCl, 20 mM TRIS and 5 mM imidazole and pH adjusted to 7.0. It was then mixed with a nickel-charged nitrilotriacetic acid cross-linked to 6% agarose resin (Ni–NTA) in a ratio of approximately 1 mL Ni–NTA to 10 mL culture supernatant and incubated for 2 h at 4 °C shaking gently. After incubation the affinity chromatography was performed in 20 mL columns. Non-bound culture supernatant was removed as flow through, and Ni–NTA resin was washed three times with 10 mL 5 mM imidazole, 2 × 7.5 mL 15 mM imidazole, 2 × 5 mL 30 mM imidazole and 1 × 5 mL 60 mM imidazole. Elution was carried out with 6 mL 500 mM imidazole. The DyP-containing elution fraction was concentrated and re-buffered in 0.1 M acetate buffer of pH 5 using ultrafiltration.
Enzyme activities
DyP and MnP activities were determined photometrically (EONTM High Perfomance Microplate Spectrophotometer, BioTek) through Mn3+ malonate formation at 270 nm (ε 270 = 11.6 mM−1 cm−1) [32]. The assay was performed with 1 mM MnSO4 in 0.1 M sodium malonate buffer of pH 4.5 at 30 °C and started by addition of 0.1 mM hydrogen peroxide. Blanks were carried out with omission of hydrogen peroxide. One enzymatic Unit (U) was defined as the amount of Mn3+ in µmol per minute which was released by the DyP or MnP at the given conditions.
Laccase activity was measured photometrically at 420 nm (ε 420 = 36.0 mM−1 cm−1) with 0.1 mM 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) (Amresco) in 0.1 M sodium malonate buffer of pH 4.5 at 30 °C [2]. One enzymatic Unit (U) was defined as the amount of ABTS radical in µmol per minute which was released by the laccase at the given conditions.
Effect of temperature and pH on Ftr-DyP activity were examined using the manganese malonate assay as described above [4]. Relative activities were calculated related to the highest activity. pH optimum and stability were determined in 0.1 M malonate buffer for the pH range of 2.5–6.0, whilst for the pH stability the Ftr-DyP was incubated for 24 h at room temperature at the respective pH prior to the activity assay. The determination of the temperature optimum was carried out in a range of 20–70 °C, whilst for the temperature stability the enzyme solution was incubated for 24 h at pH 4.5 at the respective temperature prior to enzyme activity measurement.
Kinetic constants were determined photometrically for Mn2+, ABTS, phenol red, Reactive blue 19 (RB19) and Reactive black 5 (RB5) as substrates for the Ftr-DyP. Activity assays were carried out in 0.1 M malonate buffer of pH 4.5 at 30 °C for concentrations in a range from 5 up to 750 µM and addition of 0.1 mM hydrogen peroxide. RB19 decolorization was measured at 595 nm (ε 595 = 10 mM−1 cm−1), RB 5 at 598 nm (ε 598 = 30 mM−1 cm−1) and phenol red conversion at 605 nm (ε 605 = 22.1 mM−1 cm−1) [4]. The resulting linear reaction rates were plotted against the substrate concentration, and the various K m-values were calculated using SigmaPlot 12.5 software assuming a one side saturation ligand binding to the active site of the enzyme.
Protein concentration was determined by the Lowry method. External calibration using solutions of BSA in a concentration range of 0–0.9 mg/mL in 50 mM acetate buffer of pH 5.0 was performed according to the instruction manual of the DC Assay kit (Biorad). After incubation for 15 min the absorption was measured photometrically at 750 nm and the protein concentration calculated.
(+)-Valencene conversion
Valencene conversions were carried out in 4 mL gas tight glass vials at a shaking rate of 200 rpm for 24 h at 30 °C unless indicated otherwise. Enzyme solutions buffered in 50 mM MES of pH 5.0 and a total volume of 1 mL were prepared. The reaction was started by addition of 0.5 µL (+)-valencene (Santa Cruz Biotechnology) to the aqueous phase. Enzyme activities of 5 U/mL purified Ftr-Lcc and 10 U/L purified Ftr-DyP were used, about one-fifth of the enzyme activities originally found in the culture supernatant of Funalia trogii. The reaction was performed in all combinations with and without 1 mM Mn2+ and 2.5 mM p-coumaric acid (p-CA). The culture supernatant of Funalia trogii was diluted to the same enzyme amounts and also used for conversion.
Valencene conversion was also tested using reference enzymes from other basidiomycetes, a manganese peroxidase (MnP) from Nematoloma frowardii (Sigma-Aldrich) and a recombinant DyP from Mycetinis scorodonius (MaxiBright®, TSM). The enzymes were used in isoactive activities of 10 U/L referring to the Mn2+ oxidation. The reaction was carried out under the same conditions as described before, with 1 mM Mn2+ added, with and without additional 0.2 mM hydrogen peroxide.
The effect of superoxide dismutase (SOD) (bovine, recombinant, Sigma-Aldrich) on (+)-valencene conversion with Ftr-DyP/Mn2+ was investigated using 0.6 U/L SOD, 30 U/L of Ftr-DyP and 1 mM Mn2+ in 50 mM MES buffer of pH 7.0. A biological blank was carried out using heat inactivated SOD (95 °C for 15 min).
All experiments were performed as triplicates. After incubation, valencene and its conversion products were extracted with 1 mL hexane containing 207.4 mg/L n-decanol as internal standard (IS). The extract was then dried with anhydrous sodium sulfate and subsequently measured by gas chromatography (GC). GC analyses were performed by an Agilent 7890A instrument equipped with a VF-WAX ms column (30 m × 0.25 mm id, 0.25 µm, Agilent), an on-column injection port and a flame ionisation detection (FID) system. Hydrogen was used as carrier gas at a constant flow rate of 1.2 mL per minute. One microlitre of the sample was injected by an autosampler and measured with the following method: 40 °C (3 min), a temperature ramp at a rate of 10 °C per minute until 230 °C and a final hold time of 10 min. The analytes were semi-quantified referring to the area of the internal standard. Therefore the relative response factor of (+)-nootkatone compared to the internal standard was taken into account and also used for the approximate quantification of the nootkatols. The mass concentrations were then converted into molar concentrations and molar percentage yields were calculated referring to the amount of (+)-valencene in mol determined from the blank solution.
Products were identified through GC mass spectrometric (GC–MS) analysis [Agilent 7890 B, 5977 A MSD, equipped with a VF-WAX ms column (30 m × 0.25 mm id, 0.25 µm, Agilent)] and comparison of the normal retention indices throughout both GC systems and with literature [10]. GC–MS analyses were performed with helium as carrier gas, 0.25 µL injection volume and a flow rate of 1.0 mL/min. MS scans were run in a range of m/z 34–500 with a scan rate of 3.1 scans/s. The GC temperature program was the same as in the GC-FID analyses.
To investigate whether the mechanism was based on (+)-valencene radical formation in allylic position, experiments with the addition of the spin trap 2-methyl-2-nitrosopropane were performed. MS analyses were carried out with electronic spray ionisation (ESI) in positive and negative mode (30 V/− 40 V), while detection was performed with a quadrupole analyzer in an m/z range from 140 to 500.
Results and discussion
Enzyme purification
The Funalia trogii strain was cultivated in SNL medium supplemented with wheat bran and harvested after 21 days of cultivation. The culture supernatant was used for purification of the Ftr-DyP, which consisted of two precipitation steps at 280 and 250 g/L ammonium sulfate, followed by an anion exchange chromatography and a final purification step using size exclusion chromatography. Judged by electrophoretic purity, the method was suitable to successfully purify the Ftr-DyP (Fig. 2, lane 1). A 12.2 fold purification was achieved at the cost of a low over-all recovery (Table 2). Thus, recombinant production was strived for.

SDS-PAGE analysis of the purified native Ftr-DyP (1), native Ftr-DyP with Endo H (2), recombinant Ftr-DyP (3), recombinant Ftr-DyP with Endo H (4), Endo H (5) and protein marker (M)
Amplification and expression of the Ftr-DyP
The respective protein band of the Ftr-DyP from SDS-PAGE analysis was tryptically hydrolysed and further analysed by ESI–MS/MS. Four peptides were detected (DFDTKPNPGQETVRQGVILCGR, SWAVDGSFLAFR, CPFAAHTR, GGEYFFSPSIPALR) which according to BLAST searches showed sequence homology to fungal DyPs.
The initial amplification using the primers FtrDyP_for1 and FtrDyP_rev1 yielded a 771 bp long sequence which contained the peptides SWAVDGSFIAFR and CPFAAHTR. It was concluded that the correct partial gene was amplified using the degenerated primers. The next step was to identify the 5′ and 3′ flanking sequences to obtain the whole sequence. The previously obtained DNA sequence was used to create new specific primers for the different RACEs. To identify the 3′ end a combination of the 3′ anchor primer, which bound to the 3′ adapter primer that was used for the reverse transcription, was used as well as the gene specific primer FtrDyP_for2. The PCR yielded a 470 bp long sequence verifying the peptide hit GGEYFFSPSIPALR at the end of the gene. For the 5′ RACE an adapter was ligated to the transcribed cDNA, providing a known sequence at the end of the transcripts. A first round of PCR using the primers Adp_nested_for and FtrDyP_rev2 did not yield an amplificate, which was believed to be caused by a too low concentration of successfully ligated primer-cDNA complex. Therefore, a nested PCR was performed which yielded a 756 bp long sequence comprising the rest of the missing sequence information. The sequence confirmed the presence of the last peptide hit DFDTKPNPGQETVRQGVILCGR, which gave strong evidence that the gene corresponding to the purified protein was identified. With the sequence in hand a last set of primers was designed, which was used to amplify the whole coding sequence of the gene generating a 1455 bp long fragment. When the cDNA based sequence was compared to the gDNA, six introns were identified ranging from 55 to 61 bp which was comparable to the DyP1 of the closely related Trametes versicolor (51–62 bp) [5].
The amino acid sequence of the Ftr-DyP was aligned to those of other fungal DyPs (Fig. 3). Its uniqueness became apparent on amino acid level, as the Ftr-DyP showed only low identity to the other DyPs. According to blastp searches, the Ftr-DyP was most similar to the DyPs from G. lucidum with 75% and D. squalens with 74%. Identity to the DyPs from P. ostreatus and M. scorodonius, which also converted Mn2+, was even lower.

Amino acid sequence alignment of the Ftr-DyP from Funalia trogii (Ftr, GenBank: MF953832) with DyPs from Ganoderma lucidum (Glu, GenBank: ADN05763.1), Dichomitus squalens (Dsq, GenBank: XP_007370740.1), Trametes versicolor (Tve, GenBank: XP_008039377.1), Pleurotus ostreatus (Pos, GenBank: KDQ22873.1) and Mycetinis scorodonius (Msc, GenBank: CAP53934.1)
The obtained Ftr-DyP gene was cloned into the P. pastoris expression vector pPIC9 using the appropriate primers and restriction enzymes. The initial expression of the Ftr-DyP in deep well plates resulted in average activities of about 36 U/L, while the best performing colonies produced activities up to 180 U/L, which may have resulted from a multiple insertion of the expression construct [22]. Further expression experiments were conducted using these clones for maximum protein production.
For the comparison of native and heterologous Ftr-DyP, both enzyme preparations were analysed by SDS-PAGE before and after treatment with Endo H (Fig. 2, lanes 2–5). The native Ftr-DyP as well as the recombinant Ftr-DyP after deglycosylation showed a molecular mass of around 44 kDa. Calculation from the amino acid sequence gave a molecular mass of 52.3 kDa, which was in the range reported for the majority of fungal DyPs of around 50–55 kDa [4, 26]. It remains unclear whether processing to an uncommon tertiary structure during secretion caused this difference. Treatment with endoglycosidase H prior to SDS-PAGE analysis showed that the native Ftr-DyP was not glycosylated. The recombinant Ftr-DyP, on the contrary, consisted of a mixture of pure and glycosylated enzyme species, as after deglycosylation only the lower protein band of the initial two protein bands is left and appears more intense.
Biochemical characterization
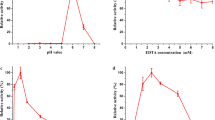
Until now only few fungal DyPs have been characterized [1, 4]. The Ftr-DyP showed a high temperature optimum at 60 °C, whereas its temperature stability decreased slowly above 30 °C (Fig. 4). During the incubation time of 24 h, the enzyme can be considered quite stable, as for the incubation at 40 °C a residual activity of about 70% remained.

The temperature optimum of the Ftr-DyP a determined for the Mn2+ conversion at pH 4.5 and temperature stability b after 24 h of incubation at the respective temperature at pH 4.5
The pH optimum of the Ftr-DyP was at pH 4.0 for phenol red as the substrate and at pH 5.0 for the Mn2+ oxidation (Fig. 5). These findings corresponded well with the reported pH optima of other fungal DyPs which ranged from pH 2 to 5 [1, 4, 23]. The enzyme was stable at the tested pH range of 2.5–6.5, while stability slightly decreased below pH 3.5, probably due to conformational changes (Fig. 5).

pH Optimum of the Ftr-DyP a determined for Mn2+ and phenol red conversion at 30 °C and pH stability of the Ftr-DyP b after 24 h incubation at room temperature and the respective pH
The substrate specificity of the Ftr-DyP was determined (Table 3). Comparing the tested substrates among each other, the Ftr-DyP showed the highest catalytic efficiency towards the synthetic peroxidase substrate ABTS with 54 ± 7 s−1 mM−1, followed by the anthraquinone dye Reactive blue 19 with 14 ± 2 s−1 mM−1. It also converted the recalcitrant azo dye Reactive black 5, but with a lower catalytic efficiency of 2.2 ± 0.4 s−1 mM−1. The Ftr-DyP also showed activity towards phenol red and Mn2+, but the catalytic efficiencies were low. The activity towards Mn2+ is remarkable, since—contrary to bacterial DyPs—fungal DyPs were thought not to catalyze the oxidation of Mn2+/Mn3+. Fernández-Fueyo et al. recently reported this property for two DyPs of P. ostreatus [3]. The same trait was also found for the Ftr-DyP and the DyP of M. scorodonius in this work. The catalytic efficiency of the Ftr-DyP towards Mn2+ was three magnitudes lower compared to those reported for MnPs, which ranged from (1–10)·103 s−1mM−1, and in this respect it resembled the Pleos-DyP1 from P. ostreatus [4, 13].
(+)-Valencene conversion
The Ftr-DyP was used to convert (+)-valencene to the grapefruit-like character-impact compound (+)-nootkatone. (+)-Valencene conversion was compared using various combinations of Ftr-DyP, Ftr-Lcc, p-CA and Mn2+ (Table 4), whereby enzyme ratios were used as originally found in the culture supernatant of F. trogii. For better understanding of the mechanism, the experiments were also performed with isoactive amounts of a DyP from M. scorodonius and a MnP from N. frowardii with Mn2+ and hydrogen peroxide as additives. References to the experiment numbers of Table 4 are given as bold numbers in parentheses.
The mixture of Ftr-Lcc with p-CA (4) led to moderate nootkatone formation with a yield of 5 mol-%. p-CA was chosen, because the presence of wheat bran in the fungal culture was supposed to result in the liberation of such phenylpropenoic moieties from the plant material. Other phenolic derivatives and ABTS were tested as mediators, but were less effective (data not shown). As the laccase was not able to fully catalyze the reaction on its own (2), the mechanism can be assumed to be mediated by p-CA, which as a phenolic substrate can be oxidized by the laccase to its phenoxy form. The mechanism of the conversion (Fig. 6) probably takes place via hydroperoxide formation of the (+)-valencene at its allylic position (C7), as was suggested by Huang et al. [9]. The mediation might proceed through a hydrogen transfer from the allylic position of (+)-valencene back to the phenoxy radical, and a valencene hydroperoxide radical is formed subsequently after covalent binding of molecular triplet oxygen. Decomposition of the hydroperoxide would result in the formation of two main stable product groups, the alcohols β- and α-nootkatol and the ketone (+)-nootkatone [9]. An increase of (+)-nootkatone yield to 20.9 mol-% was observed, when Mn2+ was added (5), while no conversion took place in the mixture of Ftr-Lcc and Mn2+ alone (3) or in the blank of p-CA with Mn2+ (1). Mn2+ cannot be oxidized by laccase as no complexing agents are present and the redox potential of Mn2+ is highly influenced by its chelation state [12, 27]. Therefore, it is more likely that the role of Mn2+ was restricted to the catalyzed in situ decomposition of the (+)-valencene peroxide, resulting in higher product yields. A similar mechanism for the decomposition of organic hydroperoxides is known for the metal catalyzed autooxidation (Fig. 6) [7, 33].

Proposed reaction sequence for the (+)-valencene conversion with the Ftr-DyP/Ftr-Lcc system and Mn2+/p-CA as mediators. R-H stands for the hydrocarbon bond of (+)-valencene in allylic position and R· for its radical form. Parallel arrows symbolize reaction paths which can take place independently from each other
Ftr-DyP converted (+)-valencene with p-CA (9), as well as with Mn2+ alone (8). As the reaction was carried out without the addition of hydrogen peroxide, an oxygen based pathway has to be assumed. In fact, additional experiments showed that hydrogen peroxide had no effect on (+)-nootkatone formation for the Ftr-DyP catalysed reaction (data not shown). The ability of peroxidases to use oxygen as electron acceptor has been described for plant peroxidases, suggesting the formation of superoxide anion radicals [25]. Furthermore, DyP activity in the absence of hydrogen peroxide was found for β-carotene degradation of M. scorodonius and B. adusta cultures [8, 18] as well as the dye-decolorizing activity of DyPs, when they were first described [11]. To investigate a possible mechanism through superoxide formation, experiments with superoxide dismutase (SOD) and Ftr-DyP were performed at 30 °C for 24 h at pH 7.0 to ensure SOD activity. While there was product formation in the Ftr-DyP/Mn2+ mixture at pH 7, addition of SOD showed a weak inhibitory effect (data not shown). This indicated that superoxide anions were transiently formed during the reaction, which contributed to the (+)-valencene oxidation. Thus, formation of superoxide anions seemed to occur when oxygen was used as substrate by the Ftr-DyP, as it was proposed for plant peroxidases [25]. p-CA or Mn2+ were presumably required to reduce the ferric ion in the heme center of the Ftr-DyP to complete the catalytic cycle, generating the phenoxy form of p-CA or Mn3+. While the reaction with the phenoxy form of p-CA might proceed similar to the Ftr-Lcc catalysed reaction, oxidation might also have occurred through Mn3+ ions, as shown for the (+)-valencene conversion with Mn3+ acetylacetonate as oxidizing agent in an aqueous system (24). The activity of the Ftr-DyP might help to reduce the required inorganic oxidant, allowing the concentration of manganese ions to be decreased down to catalytic levels which can then be oxidized to the active, but unstable +III valence in situ. Additional experiments using Ftr-DyP with 0.1 mM Mn2+ showed that it was possible to further decrease Mn2+ concentration without lowering (+)-nootkatone yields (data not shown). Mn2+ ions at a concentration of 0.16 mM are a regular constituent of the standard nutrient liquid (SNL) medium in which F. trogii was grown.
The DyP of M. scorodonius and MnP from N. frowardii did not show comparable results, even when hydrogen peroxide was provided (20–23) or the chelating malonate buffer was used. This may be explained by either low enzyme stability or a role of the Ftr-DyP beyond the provision of Mn3+.
The most efficient conversion was achieved when Ftr-DyP and Ftr-Lcc were combined in the presence of Mn2+ and p-CA (15). With this combination 99% of the (+)-valencene was converted, while (+)-nootkatone was obtained as the major volatile product with a calculated molar yield of 36% (considering GC response factors; Fig. 7). (+)-Nootkatone formation was favoured, as α-/β-nootkatols yielded only 4.3 mol-% altogether. Caused by the radical nature of the reaction, part of the (+)-valencene might have reacted to non-volatile, polymeric products, thus leading to less than 100 mol-% of the volatile products. Very similar quantitative results were achieved with a Mn2+ supplemented culture supernatant of F. trogii (18). The easy to generate culture supernatant could serve as a starting point for an industrial upscaling.

GC chromatogram of an n-hexane extract of the (+)-valencene conversion of the blank sample (a) and after incubation with the Ftr-DyP/Ftr-Lcc system and added Mn2+ and p-CA (b) after 24 h of incubation at 30 °C with (+)-valencene (1), IS (2), β-nootkatol (3) and (+)-nootkatone (4)
To investigate whether the mechanism proceeds through radical formation of (+)-valencene, experiments with the addition of the spin-trap reagent 2-methyl-2-nitrosopropane (2-MNP) were performed. The adduct formation of (+)-valencene with 2-MNP was expected. LC–MS analyses confirmed the formation of this adduct, but there was no significant increase compared to the blank preparation without enzymes. These findings do not necessarily exclude a radical mechanism, as the transient concentration of radicals might have been too low to differentiate from the high background of the blank sample.
Kinetics of nootkatone formation
Reaction kinetics of (+)-nootkatone formation was investigated for the Ftr-DyP/Ftr-Lcc mixture and the culture supernatant of F. trogii with 1 mM Mn2+ as supplement for a period of 48 h using 1 or 5 µL (+)-valencene in 1 mL reaction mixtures (Fig. 8). For the conversion of 1 µL (+)-valencene per mL, the maximum (+)-nootkatone concentration was achieved after 16 h of incubation. At this point, (+)-valencene was completely converted. Consequently, there was no further increase in (+)-nootkatone concentration with proceeding reaction time. When 5 µL/mL (+)-valencene were provided, the formation of (+)-nootkatone increased further up to 1100 mg/L after 48 h, although with a lower rate than in the beginning of the reaction. Conversion with the culture supernatant of F. trogii resulted in slightly lower initial conversion rates than in the experiments with the pure enzymatic systems. Nevertheless, the overall course of kinetics for both systems were similar. Activity measurements for both enzymes showed a remaining activity of 60–80% after 48 h at 30 °C (data not shown). The course of kinetics also showed that the ratio of (+)-nootkatone to nootkatols increased over time, especially for the 1 µL (+)-valencene reaction mixtures (data not shown). These findings indicated that nootkatols were further oxidized to (+)-nootkatone. The proceeding oxidation became more noticeable the more (+)-valencene was converted and the more nootkatols accumulated as oxidizable intermediates in the solution. Further experiments revealed that the Ftr-DyP is able to oxidize α-nootkatol to (+)-nootkatone without any required additives, while this reaction was not catalysed by the Ftr-Lcc or did proceed by oxidation with Mn3+ ions. This feature favours the product distribution towards an excess of (+)-nootkatone during (+)-valencene conversion.

Reaction kinetics of (+)-nootkatone formation with Ftr-DyP/Ftr-Lcc mixture and the culture supernatant of F. trogii with 1 mM Mn2+ at 30 °C and 200 rpm from 1 or 5 µL (+)-valencene in 1 mL reaction mixture
Conclusion
A complete substrate consumption in conversion experiments of (+)-valencene to (+)-nootkatone was achieved by the one-pot reaction catalysed by combined Ftr-DyP and Ftr-Lcc. It might be possible to increase the high (+)-nootkatone yield of 36 mol-% by optimization of reaction parameters, such as mediator or Mn2+ concentration, ratio and activity of the enzymes, and physical conditions. By an increase of the initial (+)-valencene concentration and reaction time to 48 h yields of up to 1100 mg/L (+)-nootkatone were achieved. The gap of the mass balance between consumed (+)-valencene and (+)-nootkatone/nootkatol formation indicated that high-boiling, non-GC visible oxidation products were formed, most likely through radical reactions. Mechanistic investigations will be necessary to clarify the reaction steps in this complex system. Ftr-DyP showed a broad pH-optimum, a high thermostability, a broad substrate spectrum, and catalytic activity in the absence of hydrogen peroxide. Further work will have to correlate these unique properties with the tertiary structure of the enzyme. With the discovery of more fungal DyPs able to oxidize Mn2+ ions, the exclusive association of this trait with bacterial DyPs will no longer be valid.
References
Behrens CJ, Zelena K, Berger RG (2016) Comparative cold shock expression and characterization of fungal dye-decolorizing peroxidases. Appl Biochem Biotechnol 179:1404–1417
Bourbonnais R, Paice MG (1990) Oxidation of non-phenolic substrates. FEBS Lett 267:99–102. https://doi.org/10.1016/0014-5793(90)80298-W
Esparan V, Krings U, Struch M, Berger RG (2015) A three-enzyme-system to degrade curcumin to natural vanillin. Molecules 20:6640–6653
Fernández-Fueyo E, Linde D, Almendral D, López-Lucendo MF, Ruiz-Dueñas FJ, Martínez AT (2015) Description of the first fungal dye-decolorizing peroxidase oxidizing manganese (II). Appl Microbiol Biotechnol 99:8927–8942
Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martínez AT, Otillar R, Spatafora JW, Yadav JS (2012) The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336:1715–1719
Fraatz MA, Riemer SJ, Stöber R, Kaspera R, Nimtz M, Berger RG, Zorn H (2009) A novel oxygenase from Pleurotus sapidus transforms valencene to nootkatone. J Mol Catal B Enzym 61:202–207
Hiatt RR, Mill T, Mayo FR (1968) Homolytic decompositions of hydroperoxides. I. Summary and implications for autoxidation. J Organ Chem 33:1416–1420
Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T (2010) New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol 87:871–897
Huang R, Christenson PA, Labuda IM (2001) Process for the preparation of nootkatone by laccase catalysis. US6200786B1
Kaspera R, Krings U, Nanzad T, Berger RG (2005) Bioconversion of (+)-valencene in submerged cultures of the ascomycete Chaetomium globosum. Appl Microbiol Biotechnol 67:477–483
Kim SJ, Ishikawa K, Hirai M, Shoda M (1995) Characteristics of a newly isolated fungus, Geotrichum candidum Dec 1, which decolorizes various dyes. J Ferment Bioeng 79:601–607
Kozlov YN, Zharmukhamedov SK, Tikhonov KG, Dasgupta J, Kazakova AA, Dismukes GC, Klimov VV (2004) Oxidation potentials and electron donation to photosystem II of manganese complexes containing bicarbonate and carboxylate ligands. Phys Chem Chem Phys 6:4905–4911
Kuan I-C, Johnson KA, Tien M (1993) Kinetic analysis of manganese peroxidase. The reaction with manganese complexes. J Biol Chem 268:20064–20070
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Leonhardt R-H, Berger RG (2014) Nootkatone. In: Schrader J, Bohlmann J (eds) Biotechnology of isoprenoids. Advances in Biochemical Engineering/Biotechnology, vol 148. Springer, Cham, pp 391–404
Leonhardt R-H, Plagemann I, Linke D, Zelena K, Berger RG (2013) Orthologous lipoxygenases of Pleurotus spp.—a comparison of substrate specificity and sequence homology. J Mol Catal B Enzym 97:189–195
Lin-Cereghino J, Wong WW, Giang W, Luong LT, Vu J, Johnson SD, Lin-Cereghino GP (2005) Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. Biotechniques 38:44
Linke D, Leonhardt R, Eisele N, Petersen LM, Riemer S, Nimtz M, Berger RG (2015) Carotene-degrading activities from Bjerkandera adusta possess an application in detergent industries. Bioprocess Biosyst Eng 38:1191–1199
Linke D, Matthes R, Nimtz M, Zorn H, Bunzel M, Berger RG (2013) An esterase from the basidiomycete Pleurotus sapidus hydrolyzes feruloylated saccharides. Appl Microbiol Biotechnol 97:7241–7251
Papinutti VL, Diorio LA, Forchiassin F (2003) Production of laccase and manganese peroxidase by Fomes sclerodermeus grown on wheat bran. J Ind Microbiol Biotechnol 30:157–160. https://doi.org/10.1007/s10295-003-0025-5
Rickert A, Krombach V, Hamers O, Zorn H, Maison W (2012) Enzymatic allylic oxidations with a lyophilisate of the edible fungus Pleurotus sapidus. Green Chem 14:639–644
Romanos M (1995) Advances in the use of Pichia pastoris for high-level gene expression. Curr Opin Biotechnol 6:527–533
Salvachúa D, Prieto A, Martínez ÁT, Martínez MJ (2013) Characterization of a novel dye-decolorizing peroxidase (DyP)-type enzyme from Irpex lacteus and its application in enzymatic hydrolysis of wheat straw. Appl Environ Microbiol 79:4316–4324
Sambrook J, Russell DW (eds) (2001) Molecular cloning: a laboratory manual, vol 3. Cold Spring Harbor Laboratory Press, New York
Savitsky PA, Gazaryan IG, Tishkov VI, Lagrimini LM, Ruzgas T, Gorton L (1999) Oxidation of indole-3-acetic acid by dioxygen catalysed by plant peroxidases: specificity for the enzyme structure. Biochem J 340:579–583
Scheibner M, Hülsdau B, Zelena K, Nimtz M, De Boer L, Berger RG, Zorn H (2008) Novel peroxidases of Marasmius scorodonius degrade β-carotene. Appl Microbiol Biotechnol 77:1241–1250
Schlosser D, Höfer C (2002) Laccase-catalyzed oxidation of Mn2+ in the presence of natural Mn3+ chelators as a novel source of extracellular H2O2 production and its impact on manganese peroxidase. Appl Environ Microbiol 68:3514–3521
Schulz S, Girhard M, Gaßmeyer SK, Jäger VD, Schwarze D, Vogel A, Urlacher VB (2015) Selective enzymatic synthesis of the grapefruit flavor (+)-nootkatone. ChemCatChem 7:601–604
Sprecher E (1959) Über die Guttation bei Pilzen. Planta 53:565–574. https://doi.org/10.1007/bf01937847
Struch M, Krahe N-K, Linke D, Mokoonlall A, Hinrichs J, Berger RG (2016) Dose dependent effects of a milk ion tolerant laccase on yoghurt gel structure. LWT-Food Sci Technol 65:1144–1152
Troutt AB, McHeyzer-Williams MG, Pulendran B, Nossal G (1992) Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proc Natl Acad Sci 89:9823–9825
Wariishi H, Valli K, Gold MH (1992) Manganese (II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. J Biol Chem 267:23688–23695
Waters W (1971) The kinetics and mechanism of metal-catalyzed autoxidation. J Am Oil Chem Soc 48:427–433
Wriessnegger T, Augustin P, Engleder M, Leitner E, Müller M, Kaluzna I, Schürmann M, Mink D, Zellnig G, Schwab H (2014) Production of the sesquiterpenoid (+)-nootkatone by metabolic engineering of Pichia pastoris. Metab Eng 24:18–29
Zhang X-H, Chiang VL (1996) Single-stranded DNA ligation by T4 RNA ligase for PCR cloning of 5′-noncoding fragments and coding sequence of a specific gene. Nucleic Acids Res 24:990–991
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kolwek, J., Behrens, C., Linke, D. et al. Cell-free one-pot conversion of (+)-valencene to (+)-nootkatone by a unique dye-decolorizing peroxidase combined with a laccase from Funalia trogii . J Ind Microbiol Biotechnol 45, 89–101 (2018). https://doi.org/10.1007/s10295-017-1998-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-017-1998-9