
Abstract
Polylactic acid (PLA) is an important renewable polymer, but current processes for producing its precursor, lactic acid, suffer from process inefficiencies related to the use of bacterial hosts. Therefore, improving the capacity of Saccharomyces cerevisiae to produce lactic acid is a promising approach to improve industrial production of lactic acid. As one such improvement required, the lactic acid tolerance of yeast must be significantly increased. To enable improved tolerance, we employed an RNAi-mediated genome-wide expression knockdown approach as a means to rapidly identify potential genetic targets. In this approach, several gene knockdown targets were identified which confer increased acid tolerance to S. cerevisiae BY4741, of which knockdown of the ribosome-associated chaperone SSB1 conferred the highest increase (52 %). This target was then transferred into a lactic acid-overproducing strain of S. cerevisiae CEN.PK in the form of a knockout and the resulting strain demonstrated up to 33 % increased cell growth, 58 % increased glucose consumption, and 60 % increased l-lactic acid production. As SSB1 contains a close functional homolog SSB2 in yeast, this result was counterintuitive and may point to as-yet-undefined functional differences between SSB1 and SSB2 related to lactic acid production. The final strain produced over 50 g/L of lactic acid in under 60 h of fermentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabolic engineering can enable the high level production of a variety of interesting bio-based chemicals and polymers [37]. Among these, polylactic acid (PLA) is increasingly recognized as a sustainable alternative to petroleum-based plastics, and has seen wide use in various applications due to its desirable material properties and biodegradability. Bioproduction of PLA begins with lactic acid, which is currently produced in large quantities in carbohydrate fermentations by lactic acid bacteria (LAB) [15]. After fermentation, two molecules of lactic acid can be further hydrated to lactide and subsequently polymerized into polylactic acid. Lactate production through bacterial fermentation is well known in the so-called lactic acid bacteria (LAB) such as Lactobacillus pentosus [7], Lactobacillus brevis [8], and Bacillus coagulans [5, 29]. B. coagulans has been reported to produce lactate with as high as 99.6 % yield, 215.7 g/L titer, and 4.0 g/(L*h) productivity during fed-batch fermentation using xylose [43]. Recent studies have reported lactate production from lignocellulosic biomass, but LA titer and yield are lower than that from pure sugar. In particular, LA production by B. coagulans LA204 using agricultural stover showed 68 % yield, 97.59 g/L titer, and 1.63 g/(L*h) productivity [21]. Additionally, Pediococcus acidilactici DQ2 has been shown to produce lactate from sulfuric acid-treated corn stover at 77.2 % yield, 101.9 g/L titer, and 1.06 g/(L*h) productivity [44].
However, bacterial fermentations of lactic acid suffer from a variety of process disadvantages, including the necessity for high fermentation temperatures (which increases energy input), the need for pH neutralization (resulting in high salt content), and the possibility of phage contamination (which can halt a fermentation and incur substantial sterilization costs [18]). As an alternative, the yeast Saccharomyces cerevisiae is an excellent host for the production of a diverse array of small molecules with relevance as fuels, chemicals, and therapeutics due to its preference for lower fermentation temperatures and resistance to contamination [27]. Unlike LAB, yeast is not a native producer of lactic acid and therefore must be genetically engineered to produce and become tolerant to this compound.
Metabolically engineered yeasts which overexpress exogenous L-lactate dehydrogenase (L-LDH) were first reported by Dequin and Barre [16]. In these strains, yeast converts glucose to both ethanol and lactate. To focus metabolic flux toward the production of lactate, the disruption of ethanol fermentation is essential. Ethanol fermentation is mainly dependent on the activity of pyruvate decarboxylase (PDC), which converts pyruvic acid into acetaldehyde [33]. Deletion of genes encoding PDC, in combination with heterologous overexpression of L-LDH, results in engineered strains capable of producing lactic acid with little or no ethanol formation [1, 23, 32, 35, 39]. There are a few reports of high lactic acid titers in specific yeast strains. In particular, a S. cerevisiae strain engineered for improved intracellular redox balance was found to produce 117 g/L of lactate at a yield of 58 % in a fed-batch bioreactor under low pH conditions [26]. The highest LA titer was reported from S. cerevisiae using a cane juice-based medium at a pH of 5.2 (122 g/L lactate, 61 % yield) [35], which compares favorably with that of other yeasts such as Candida sonorensis (92 g/L lactate, 94 % yield under neutralizing conditions) [22]. Currently, a large quantity of neutralizing agents such as CaCO3, NaOH, and NH4OH are added during fermentation to reduce the inhibitory effects of acidity on cell growth and overall titer. The development of an acid-tolerant strain is therefore required to improve lactic acid production at low pH and concomitantly decrease process costs due to neutralizing agents.
Traditional metabolic engineering approaches are often informed by detailed models of metabolism and algorithms which computationally search the genomic landscape for promising engineering targets [12]. However, even for well-studied organisms such as S. cerevisiae, regulatory interactions, which play critical roles in shaping complex phenotypes such as tolerance, are largely unknown, thus precluding the rational development of increased tolerance using genome-scale models and thus necessitating more high-throughput approaches. Several techniques currently exist for altering gene expression in yeast, including the use of various promoters (both constitutive and inducible [14]), RNA-based control modules [9], and gene knockout techniques [19]. Although these tools are critical to the implementation of rationally predicted genetic modifications, these approaches cannot be implemented in a high-throughput manner to discover novel targets. In addition, although collections of yeast strains have been created, which collectively contain each single gene knockout [41], utilizing these databases to multiplex genetic targets or transfer targets to alternative yeast strains is a labor-intensive process. Finally, although techniques such as random mutagenesis [11], transposon insertion libraries [25], and global transcription machinery engineering [4] are effective for genome-wide yeast engineering, these techniques are not explicitly tunable and (for the case of random mutagenesis and transposon insertion libraries) difficult to implement in polyploid hosts. Therefore, a high-throughput tunable approach to rapidly prototype knockdowns relevant to a phenotype of interest may increase the capacity to obtain transferable phenotypic effectors and could improve understanding of the yeast interactome.
To enable this capacity, we have previously developed and implemented an effective method for gene knockdown in yeast through RNA interference [13]. This approach enables targeted knockdown of endogenous gene expression by nearly 95 % without the requirement for labor-intensive genomic editing. Furthermore, this method is applicable to several different strains of S. cerevisiae and thus enables facile multiplexing of gene knockdowns across a wide variety of strains. This approach, therefore, allows for “rapid prototyping” of strain modifications to quickly identify routes for attainment of a metabolic engineering objective. This initial implementation of RNA interference in yeast can furthermore be expanded to enable a high-throughput approach for generation of knockdown phenotypes on a genome-wide scale [36]. In this scheme, knockdown targets may be identified in the absence of detailed knowledge of host metabolism. As a further benefit, this high-throughput approach would enable rapid discovery of novel gene regulatory pathways related to the phenotype of interest through the analysis of genetic targets identified from this genome-wide search.
In this work, we hypothesized that a genome-wide search for downregulation targets using RNAi could be used to improve yeast lactic acid tolerance. To this end, we constructed a genome-wide knockdown library in S. cerevisiae and, after a single round of screening, identified three knockdown targets (ssb1, adh1, and rpl41b) which increase lactic acid tolerance of S. cerevisiae BY4741 by 52, 38, and 13 %, respectively. We then transferred one of these targets (ssb1) in the form of a knockout in an l-lactic acid-overproducing strain of yeast and observed a 60 % increase in lactic acid production.
Materials and methods
Strains and media
Yeast expression vectors were propagated in Escherichia coli DH10β. E. coli strains were routinely cultivated in LB medium (Teknova, Hollister, CA, USA) at 37 °C with 225 rpm orbital shaking. LB was supplemented with 100 µg/mL ampicillin (Sigma, St. Louis, MO, USA) when needed for plasmid maintenance and propagation. Yeast strains were cultivated on a yeast synthetic complete (YSC) medium containing 6.7 g of Yeast Nitrogen Base/liter (BD Difco, Franklin Lakes, NJ, USA), 20 g glucose/L, and a mixture of appropriate nucleotides and amino acids (YSC, MP Biomedicals, Solon, OH, USA). All components were supplemented with 1.5 % agar for solid media.
For E. coli transformations, 50 µL of electrocompetent E. coli DH10β was mixed with 30 ng of ligated DNA and electroporated (2 mm Electroporation Cuvettes (Bioexpress, Kaysville, UT, USA) with Bio-Rad Genepulser Xcell (Bio-Rad, Hercules, CA, USA) at 2.5 kV. Transformants were rescued for one hour at 37 °C in 1 mL SOC Buffer (Cellgro, Manassas, VA, USA), plated on LB agar, and incubated overnight. Single clones were amplified in 5 mL LB medium and incubated overnight at 37 °C. Plasmids were isolated (QIAprep Spin Miniprep Kit, Qiagen, Valencia, CA, USA) and confirmed by sequencing.
For yeast transformations, 50 µL of chemically competent S. cerevisiae BY4741 was transformed with 1 µg of each appropriate purified plasmid according to the established protocols [19], plated on the appropriate medium, and incubated for three days at 30 °C. Single colonies were picked into 1 mL of the appropriate medium and incubated at 30 °C. Plasmids were isolated using a yeast miniprep kit (Yeast Miniprep Kit II, Zymo Research Corporation).
Ligation cloning procedures
PCRs were performed with Q5 Hot-Start DNA Polymerase (NEB, Ipswich, MA, USA) according to manufacturer’s specifications. Digestions were performed according to manufacturer’s instructions, with digestions close to the end of a linearized strand running overnight and digestions of circular strands running for 1 h at 37 °C. PCR products and digestions were cleaned with a QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA). Phosphatase reactions were performed with Antarctic Phosphatase (NEB, Ipswich, MA, USA) according to manufacturer’s instructions and heat-inactivated for 15 min at 65 °C. Ligations (T4 DNA Ligase, Fermentas, Waltham, MA, USA) were performed overnight at 22 °C followed by heat inactivation at 65 °C for 20 min. Detailed cloning procedures are described in Online Resource 1.
Recombination cloning in yeast
1 µg of each PCR fragment was digested with DpnI and cotransformed into S. cerevisiae BY4741 according to the procedure described in Hegemann and Heick. [19]. This transformation mixture was then plated on the appropriate dropout medium and allowed to grow for 3 days at 30 °C. Yeast colonies from this plate were scraped and plasmids were extracted (Zymoprep Yeast Miniprep Kit, Zymo Research, Irvine, CA, USA). This plasmid mixture was then transformed into E. coli DH10β and plated. Individual colonies were then amplified in liquid culture and plasmids were extracted. Correctly assembled plasmids were confirmed through restriction digestion and sequencing.
Growth rate analysis
Three biological replicates of each yeast strain were precultured in an orbital shaking incubator at 30 °C for 2 days in 1 mL YSC −Leu −Trp, after which they were pelleted, washed, and used to inoculate a 2 mL YSC −Leu −Trp culture containing 15 g/L lactic acid at OD600nm = 0.1. These cultures were then incubated in a rotary drum at 30 °C and optical density was measured every 24 h. All knockdown experiments utilized S. cerevisiae BY4741 ∆TRP1 for plasmid maintenance, and all knockout experiments utilized S. cerevisiae BY4741 to enable growth in tryptophan dropout media.
cDNA library generation
Total RNA was extracted from yeast using the RNA Extraction Kit (Ambion, Austin, TX, USA) and converted to cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s instructions with the exception that primer oligodTEcoRIR2 was substituted for the random hexamer primer provided with the kit. This cDNA was then purified using the Qiagen PCR cleanup kit and ligated to primer RNALigAd using T4 RNA Ligase (NEB, Ipswich, MA, USA) according to manufacturer’s instructions. This ligation was purified using a Qiagen PCR cleanup kit and amplified using Q5 hot-start DNA polymerase and primers XmaIFlankF and EcoRIFlankR2 according to manufacturer’s instructions. Amplicons ranging in size from 500 bp to 5 kb were gel-extracted (Genejet Gel Purification Kit, Life Technologies, Carlsbad, CA, USA) and re-purified using the Qiagen PCR Cleanup Kit. This purified, double-stranded, full-length cDNA was then sheared using a Covaris sonicator to an average length of 200 or 400 bp, blunt-ended, and phosphorylated using the End-It DNA End Repair Kit (Epicenter, Madison, WI, USA), and ligated to p414-GPD-rad9-MCS-rad9’-TEF’ and p414-CYC-rad9-MCS-rad9’-TEF’. These vectors result in either weak or strong expression of the downregulation cassette, thus enabling low or high levels of downregulation. This procedure thus generated 4 libraries, each of which contained over 105 distinct members and thus enabled good coverage of the yeast transcriptome.
Screening and target identification strategy
Yeast expressing each knockdown library was used to inoculate 100 mL cultures containing various quantities of lactic acid at an initial optical density of 0.1. These screening libraries were then allowed to grow until they reached an optical density of greater than one, at which point they were subcultured at a 1:100 ratio to a fresh culture with an increased concentration of lactic acid. This process was repeated three times to generate four enriched collections of downregulation cassettes. Finally, isolated members of these enriched collections were sequenced at random to identify targets responsible for the improved phenotype.
Development of l-lactic acid-producing yeast strains
The recombinant strain S. cerevisiae CEN.PK2 “Control” was developed for l-lactic acid production in S. cerevisiae using standard molecular techniques. Specifically, the Ura3-blaster genetic disruption method was applied for gene deletion S. cerevisiae CEN.PK2 [2]. First, the LDH gene was integrated at the pdc1 locus under the control of the PGK1 promoter by homologous recombination. Then, two more copies of LDH were introduced into cyb2 and gpd1 under the control of the CCW12 promoter by the same method, producing the “Control” strain (pdc1Δ::P PGK1 -ldh cyb2Δ::P PGK1 -ldh gpd1Δ::P CCW12 -ldh, Lee et al. 2015). To delete ssb1 in this strain, a deletion cassette with homologous flanking regions was amplified by PCR from pUC-URA3 [26] and integrated at the ssb1 locus, producing S. cerevisiae CEN.PK2 “ssb1 KO.” Genetic modifications were verified by PCR using primer sets within the flanking genomic sequence.
l-Lactic acid production
The lactic acid-producing strains (“Control” and “ssb1 KO”) were grown overnight at 30 °C, 230 rpm. The seed cultures were then transferred into a 125 mL flask containing YPD medium with 6–8 % glucose. The initial OD600nm was set to 0.5 and the fermentation condition was maintained at 30 °C, 90 rpm. For pH control experiments, calcium carbonate was added (2 g/L) to the culture. Samples were collected at 24 and 48 h for analysis.
HPLC analysis
Fermentation samples were analyzed by high-performance liquid chromatography (HPLC) for l-lactic acid, glucose, and ethanol using a Waters e2695 Separation Module instrument equipped with a Waters 2414 Differential Refractometer and a Waters 2998 Photodiode Array Detector (Waters, Milford, MA), a Fast Juice Column (50 × 7.8 mm; Phenomenex, Torrance, CA) and a Fast Acid Analysis Column (100 × 7.8 mm; Bio-Rad, Hercules, CA, USA) or, alternatively, a Fast Acid Analysis Column (100 × 7.8 mm; Bio-Rad, Hercules, CA, USA) and an Aminex HPX-87H Organic Acid Analysis Column (300 × 7.8 mm; Bio-Rad, Hercules, CA, USA). Samples were equilibrated with 2.5 mM H2SO4 in water at 60 °C, and samples were eluted with 2.5 mM H2SO4 in water at a 0.5 mL/min flow rate. Data were acquired using the Waters Millennium software.
qPCR
For each tested variant, a replicate was grown to an optical density of 0.5 and its RNA was extracted (Quick-RNA Miniprep, Zymo Research Corporation). 2 µg RNA was reverse-transcribed (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems) and quantified in triplicate (SYBR Green PCR Master Mix, Life Technologies) immediately after RNA extraction. SSB1 and SSB2 transcript levels were measured relative to that of a housekeeping gene (ALG9) (Viia 7 Real Time PCR Instrument, Life Technologies) using primers which amplified both SSB1 and SSB2 cDNA. Primers used for quantification are listed in Online Resource 1.
Supplementary methods
For additional information regarding strain construction and the identities of strains used in each figure of this paper, see Online Resource 1.
Results and discussion
Lactic acid inhibits yeast growth
To investigate the toxicity of lactic acid to our screening host, cell growth of S. cerevisiae BY4741 expressing the RNAi machinery along with a blank downregulation cassette was measured in varying quantities of lactic acid. We observed that lactic acid concentrations greater than roughly 5 g/L severely limited the growth of wild-type S. cerevisiae and thus, provided a good starting point to identify knockdown variants with improved tolerance (Fig. 1).

Growth inhibition of BY4741 by lactic acid. Wild-type BY4741 was grown in media containing various concentrations of lactic acid (0, 3, 6, 9, 12 g/L) and OD600nm was measured on day 2. The lactic acid concentration at which OD600nm was inhibited by 50 % was 5.3 g/L
Construction of cell-wide downregulation libraries
Full-length, double-stranded cDNA libraries were generated through a slight modification of established techniques [38] (see “Materials and methods”), sheared to either 200 or 400 bp in length, and ligated to a vector containing convergent promoters, a design strategy which has been shown to enable downregulation [36]. In this experiment, we utilized vectors which drove either weak or strong expression of the downregulation cassette, thus enabling low or high levels of downregulation. This procedure thus generated four libraries, each of which contained over 105 distinct members and thus enabled good coverage of the yeast transcriptome. These libraries were then screened through serial subculture in increasing lactic acid concentrations, and knockdown cassettes from each subculture were identified through Sanger sequencing of randomly isolated colonies.
Identification of knockdown targets improving lactic acid tolerance
Genome-wide knockdown libraries were generated and screened for growth in inhibitory concentrations of lactic acid, after which knockdown targets were identified through sequencing (see “Materials and methods”). These putative targets were then retransformed (along with the appropriate RNAi machinery) into a fresh strain of yeast and grown in the presence of lactic acid to confirm improvements to growth. Based on measurements of culture optical density, we observed that several knockdown cassettes, including multiple isolates targeting ADH1, SSB1/2, and RPL41B, improved tolerance to lactic acid by 38, 52, and 13 %, respectively (Fig. 2, Table 1). ADH1 encodes alcohol dehydrogenase, SSB1/2 ribosome-associated chaperones, and RPL41B a particularly small component of the 60 s ribosomal subunit. Although each of these targets represented an interesting launching point for improvement of lactic acid tolerance and elucidation of tolerance-enhancing gene networks, we delved more deeply into SSB1/2 knockdown as its improvement to lactic acid tolerance was highest.

Effects of expressing ADH1, SSB1/2, and RPL41B knockdown cassettes on yeast cell growth in lactic acid (a ADH1, b SSB1/2, c RPL41B). Yeast strains expressing knockdown cassettes were grown in 15 g/L lactic acid and OD600nm was measured each day. Error bars represent standard error of the mean among three biological replicates
SSB1 and SSB2 are close homologs arising from the genome duplication event in yeast, with 97 % nucleotide and 99 % amino acid identity to each other, respectively. Their strong similarity implies that any SSB1-targeted knockdown cassette likely also downregulates the expression of SSB2 and vice versa. We therefore denote targeting downregulation cassettes as SSB1/2 to emphasize this potential. Ssb1p and Ssb2p are ribosome-associated ATPases in the Hsp70 family and are hypothesized to act as chaperones at the ribosome exit tunnel [10, 30, 31]. Ssb1p has been shown to interact with 3315 yeast proteins, the highest of any yeast chaperone, and to share 1027 interactors with Ssb2p, which interacts with 1236 [17]. Additionally, both proteins exist in the top 5 % of yeast’s most abundant proteins [40]. It has been shown that knockout of either individual protein confers no growth defect as measured by optical density [10], whereas, knockout of both genes confers slow growth, cation sensitivity, and hypersensitivity to aminoglycoside antibiotics, presumably due to their important role in translation [24]. Despite their annotation as heat-shock proteins, their expression is considerably repressed (by up to 80 %) upon heat shock. These genes thus represented intriguing knockdown targets for improving lactic acid tolerance, as their high abundance and large interactome likely enable the alteration of complex phenotypes upon downregulation.
SSB1 in lactic acid production
We wished to examine the behavior of yeast expressing SSB1/2 knockdown cassettes in inhibitory concentrations of lactic acid. Expression of each previously identified SSB1/2 knockdown cassette yielded improvements to yeast cell growth in the presence of 15 g/L lactic acid versus a yeast strain expressing a blank vector lacking an RNAi-targeting sequence (Fig. 3a). However, we did not observe a similar increase to tolerance during ssb1 knockout, with growth rates between wild-type yeast (BY4741) and the ssb1 knockout remaining similar (Fig. 3b). Intriguingly, total SSB1/2 transcript levels in the tolerant knockdown strain were intermediate between wild-type and knockout (Fig. 4), indicating the presence of a local optimum in BY4741 lactic acid tolerance as a function of SSB1 gene expression.

Effects of a SSB1/2 knockdown or b SSB1 knockout on yeast cell growth in lactic acid. Yeast strains expressing SSB knockdown cassettes were grown in 15 g/L lactic acid and OD600nm was measured each day. Error bars represent standard error of the mean among three biological replicates

SSB1 transcript levels in engineered BY4741 strains. Yeast strains expressing the RNAi machinery only, RNAi machinery, and the SSB1 knockdown cassette, or containing a genomic knockout of SSB1 were grown in either minimal media (YSC) or minimal media containing 15 g/L lactic acid, and SSB1/2 transcripts were measured via qPCR. Error bars represent standard deviation among three technical replicates
The difference between knockdown and knockout notwithstanding, we chose to explore the utility of modifying the SSB1 locus through transferring this target as a knockout into a lactic acid-overproducing strain. This strain contains a pdc1 deletion to reduce ethanol production. In addition, CYB2, which encodes l-lactate cytochrome-c oxidoreductase, was also removed to prevent l-lactic acid degradation. In agreement with previous results, deletion of the two genes encoding glycerol-3-phosphate dehydrogenase (GPD1 and GPD2) to reduce glycerol production led to severe side effects, including poor growth and decreased tolerance toward osmotic, heat, freezing/thawing, and oxidative stresses [3, 6, 20, 28]. To engineer lactate producing strains with low glycerol production, we deleted the gene for Gpd1p, which is known to be responsible for the majority of glycerol production [34]. GPD1 deletion was sufficient to reduce glycerol production to an undetectable level without significant side effects in this study. Each of these three targets was deleted via replacement with LDH [26].
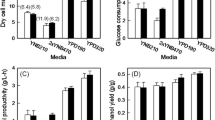
In agreement with prior results, when SSB1 was knocked out in this lactic acid-producing strain, we did not observe any improvement in tolerance toward lactic acid during growth on solid media. Remarkably though, ssb1 knockout showed a positive effect on shake flask production of l-lactic acid and increased l-lactic acid titer by 33 % to 26.21 g/L (Fig. 5a). To further test the effect of ssb1 knockout under neutralizing conditions, we introduced pH control using 2 g/L calcium carbonate. Under this condition, glucose was consumed more rapidly, so we increased the starting glucose concentration to 80 g/L for this experiment. It has been previously reported that high extracellular glucose concentrations decrease glucose transport capacity, resulting in lower overall glucose consumption [42]. However, under this pH-controlled condition, glucose consumption was dramatically increased by 58 % to 69.13 g/L, cell growth was improved by 33 % (reaching a final OD600nm of 4.02 in the knockout strain), and l-lactic acid production was enhanced by 60 % to 50.57 g/L (Fig. 5b) relative to the wild-type strain under the same conditions. Fermentations were also performed in 2.0 L jar fermenter using a defined media with pH and glucose feeding control, and the results were consistent with shake flasks (data not shown). Thus, the ssb1 knockout proved to increase the overall production of lactic acid by yeast.

Production of l-lactic acid in SSB1 knockout CEN.PK2 increased by 60 % compared to its parent strain. A flask-based fermentation test was performed in YPD medium containing 60 g/L of glucose (a) or 80 g/L of glucose (b). In b 2 g/L of calcium carbonate was added to maintain pH control. Error bars represent standard deviation among biological replicates
Through this work, we uncovered three novel knockdown targets (ADH1, SSB1, and RPL41B) which increase lactic acid tolerance in yeast. Due to its 52 % improvement to lactic acid tolerance in BY4741, we conducted a further study on the effect of ssb1 knockout for improvement of a lactic-acid-overproducing strain of yeast. Intriguingly, we observed that ssb1 knockout increased lactic acid production by 60 % in this engineered strain despite having no observable effect on lactic acid tolerance for either strain. Due to their ability to exert influence on a myriad of cellular processes, their linkage to a complex phenotype such as lactic acid tolerance is not entirely unanticipated. We found that combined SSB1/2 expression upon SSB1 knockdown was intermediate between that of wild-type and ΔSSB1 genotypes in BY4741, which was surprising as we expected that the minimal nucleotide differences between SSB1 and SSB2 would cause RNAi to reduce combined SSB1/2 expression beyond that conferred by a single ssb1 knockout alone. This result indicates that RNAi-mediated downregulation of SSB1/2 was weaker than expected, and could also indicate interesting regulatory behavior buffering levels of SSB1/2 in the knockdown. Nevertheless, this gene remains an interesting target for increased lactic acid production and may be a potential target to be explored in the engineering of tolerance to additional acids by yeast.
Conclusion
In this work, we successfully implemented a synthetic RNAi pathway to enable high-throughput identification of knockdown targets conferring increased tolerance to lactic acid. One significant advantage of the RNAi approach to gene knockdowns is the facile generation of a genome-wide knockdown library, in which each member of the library (which can be derived from cDNA or gDNA) specifically downregulates a different gene. Furthermore, this approach enables sequence-specific downregulation regardless of genomic copy number, enabling trivial downregulation of multicopy genes. Through this work, we discovered a knockdown target (SSB1) which illustrates these unique capacities of RNA interference for the discovery of genes which would otherwise be missed using more traditional knockout library-based approaches. This target increased the tolerance of yeast to lactic acid and the transfer of this target to a production host for lactic acid increased production by 60 % to over 50 g/L of lactic acid in under 60 h of fermentation.
Abbreviations
- RNAi:
-
RNA interference
- LAB:
-
Lactic acid bacteria
- PLA:
-
Polylactic acid
- L-LDH:
-
l-Lactate dehydrogenase
- PDC:
-
Pyruvate decarboxylase
- SSB1:
-
Yeast cytoplasmic chaperone
- RPL14B:
-
Yeast ribosomal 60S subunit L41B
- ADH1:
-
Yeast alcohol dehydrogenase
- YSC:
-
Yeast synthetic complete medium
- HPLC:
-
High-performance liquid chromatography
- CYB2:
-
Yeast l-lactate cytochrome-c oxidoreductase
- GPD1:
-
Yeast glycerol-3-phosphate dehydrogenase
- PGK1:
-
Yeast phosphoglycerate kinase
- CCW12:
-
Yeast cell wall mannoprotein
References
Adachi E, Torigoe M, Sugiyama M, Nikawa J-I, Shimizu K (1998) Modification of metabolic pathways of Saccharomyces cerevisiae by the expression of lactate dehydrogenase and deletion of pyruvate decarboxylase genes for the lactic acid fermentation at low pH value. J Ferment Bioeng 86:284–289. doi:10.1016/S0922-338X(98)80131-1
Alani E, Cao L, Kleckner N (1987) A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics 116:541–545
Aldiguier AS, Alfenore S, Cameleyre X, Goma G, Uribelarrea JL, Guillouet SE, Molina-Jouve C (2004) Synergistic temperature and ethanol effect on Saccharomyces cerevisiae dynamic behaviour in ethanol bio-fuel production. Bioprocess Biosyst Eng 26:217–222. doi:10.1007/s00449-004-0352-6
Alper H, Moxley J, Nevoigt E, Fink GR, Stephanopoulos G (2006) Engineering yeast transcription machinery for improved ethanol tolerance and production. Science 314:1565–1568. doi:10.1126/science.1131969
Bischoff KM, Liu S, Hughes SR, Rich JO (2010) Fermentation of corn fiber hydrolysate to lactic acid by the moderate thermophile Bacillus coagulans. Biotechnol Lett 32:823–828. doi:10.1007/s10529-010-0222-z
Blomberg A, Adler L (1989) Roles of glycerol and glycerol-3-phosphate dehydrogenase (NAD +) in acquired osmotolerance of Saccharomyces cerevisiae. J Bacteriol 171:1087–1092
Bustos G, Moldes AB, Cruz JM, Dominguez JM (2005) Influence of the metabolism pathway on lactic acid production from hemicellulosic trimming vine shoots hydrolyzates using Lactobacillus pentosus. Biotechnol Prog 21:793–798. doi:10.1021/bp049603v
Chaillou S, Bor YC, Batt CA, Postma PW, Pouwels PH (1998) Molecular cloning and functional expression in lactobacillus plantarum 80 of xylT, encoding the D-xylose-H+ symporter of Lactobacillus brevis. Appl Environ Microbiol 64:4720–4728
Chang AL, Wolf JJ, Smolke CD (2012) Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr Opin Biotechnol 23:679–688. doi:10.1016/j.copbio.2012.01.005
Craig EA, Jacobsen K (1985) Mutations in cognate genes of Saccharomyces cerevisiae hsp70 result in reduced growth rates at low temperatures. Mol Cell Biol 5:3517–3524
Crook N, Alper HS (2012) Classical strain improvement. In: Patnaik R (ed) Engineering complex phenotypes in industrial strains. Wiley, Hoboken, NJ, pp 1–33. doi: 10.1002/9781118433034.ch1
Crook N, Alper HS (2013) Model-based design of synthetic, biological systems. Chem Eng Sci 103:2–11. doi:10.1016/j.ces.2012.12.022
Crook NC, Schmitz AC, Alper HS (2013) Optimization of a yeast RNA interference system for controlling gene expression and enabling rapid metabolic engineering. ACS Syn Bio 3:307–313. doi:10.1021/sb4001432
Da Silva NA, Srikrishnan S (2012) Introduction and expression of genes for metabolic engineering applications in Saccharomyces cerevisiae. FEMS Yeast Res 12:197–214. doi:10.1111/j.1567-1364.2011.00769.x
Datta R, Tsai S-P, Bonsignore P, Moon S-H, Frank JR (1995) Technological and economic potential of poly(lactic acid) and lactic acid derivatives. FEMS Microbiol Rev 16:221–231. doi:10.1016/0168-6445(94)00055-4
Dequin S, Barre P (1994) Mixed lactic acid-alcoholic fermentation by Saccharomyces cerevisiae expressing the Lactobacillus casei L(+)-LDH. Bio/technology (Nature Publishing Company) 12:173–177
Gong Y, Kakihara Y, Krogan N, Greenblatt J, Emili A, Zhang Z, Houry WA (2009) An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol Syst Biol 5:275. doi:10.1038/msb.2009.26
Grath SM, van Sinderen D (eds) (2007) Bacteriophage: Genetics and molecular biology. Caister Academic Press, Norfolk
Hegemann JH, Heick SB (2011) Delete and repeat: a comprehensive toolkit for sequential gene knockout in the budding yeast Saccharomyces cerevisiae. In: Williams JA (ed) Methods in Molecular Biology., vol 765. Humana Press, New York, pp 189–206. doi:10.1007/978-1-61779-197-0_12
Hohmann S (2002) Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev MMBR 66:300–372
Hu J, Zhang Z, Lin Y, Zhao S, Mei Y, Liang Y, Peng N (2015) High-titer lactic acid production from NaOH-pretreated corn stover by Bacillus coagulans LA204 using fed-batch simultaneous saccharification and fermentation under non-sterile condition. Bioresour Technol 182:251–257. doi:10.1016/j.biortech.2015.02.008
Ilmen M, Koivuranta K, Ruohonen L, Rajgarhia V, Suominen P, Penttila M (2013) Production of l-lactic acid by the yeast Candida sonorensis expressing heterologous bacterial and fungal lactate dehydrogenases. Microb Cell Fact 12:53. doi:10.1186/1475-2859-12-53
Ishida N, Saitoh S, Onishi T, Tokuhiro K, Nagamori E, Kitamoto K, Takahashi H (2006) The effect of pyruvate decarboxylase gene knockout in Saccharomyces cerevisiae on l-lactic acid production. Biosci Biotechnol Biochem 70:1148–1153. doi:10.1271/bbb.70.1148
Kim SY, Craig EA (2005) Broad sensitivity of Saccharomyces cerevisiae lacking ribosome-associated chaperone ssb or zuo1 to cations, including aminoglycosides. Eukaryot Cell 4:82–89. doi:10.1128/ec.4.1.82-89.2005
Kumar A, Snyder M (2001) Genome-wide transposon mutagenesis in yeast. In: Lundblad V, Struhl K (eds) Current protocols in molecular biology, chap 13. Wiley, Hoboken, NJ. doi: 10.1002/0471142727.mb1303s51
Lee JY, Kang CD, Lee SH, Park YK, Cho KM (2015) Engineering cellular redox balance in Saccharomyces cerevisiae for improved production of l-lactic acid. Biotechnol Bioeng 112:751–758. doi:10.1002/bit.25488
Liu L, Redden H, Alper HS (2013) Frontiers of yeast metabolic engineering: diversifying beyond ethanol and Saccharomyces. Curr Opin Biotechnol 24:1023–1030. doi:10.1016/j.copbio.2013.03.005
Luyten K, Albertyn J, Skibbe WF, Prior BA, Ramos J, Thevelein JM, Hohmann S (1995) Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. EMBO J 14:1360–1371
Patel MA, Ou MS, Harbrucker R, Aldrich HC, Buszko ML, Ingram LO, Shanmugam KT (2006) Isolation and characterization of acid-tolerant, thermophilic bacteria for effective fermentation of biomass-derived sugars to lactic acid. Appl Environ Microb 72:3228–3235. doi:10.1128/aem.72.5.3228-3235.2006
Peisker K, Chiabudini M, Rospert S (2010) The ribosome-bound Hsp70 homolog Ssb of Saccharomyces cerevisiae. Biochim Biophys Acta 1803:662–672. doi:10.1016/j.bbamcr.2010.03.005
Pfund C, Lopez-Hoyo N, Ziegelhoffer T, Schilke BA, Lopez-Buesa P, Walter WA, Wiedmann M, Craig EA (1998) The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J 17:3981–3989. doi:10.1093/emboj/17.14.3981
Porro D, Bianchi MM, Brambilla L, Menghini R, Bolzani D, Carrera V, Lievense J, Liu C-L, Ranzi BM, Frontali L, Alberghina L (1999) Replacement of a metabolic pathway for large-scale production of lactic acid from engineered yeasts. Appl Environ Microb 65:4211–4215
Pronk JT, Yde Steensma H, Van Dijken JP (1996) Pyruvate metabolism in Saccharomyces cerevisiae. Yeast (Chichester, England) 12:1607–1633. doi:10.1002/(SICI)1097-0061(199612)12:16<1607:AID-YEA70>3.0.CO;2-4
Remize F, Cambon B, Barnavon L, Dequin S (2003) Glycerol formation during wine fermentation is mainly linked to Gpd1p and is only partially controlled by the HOG pathway. Yeast (Chichester, England) 20:1243–1253. doi:10.1002/yea.1041
Saitoh S, Ishida N, Onishi T, Tokuhiro K, Nagamori E, Kitamoto K, Takahashi H (2005) Genetically engineered wine yeast produces a high concentration of l-Lactic acid of extremely high optical purity. Appl Environ Microb 71:2789–2792. doi:10.1128/aem.71.5.2789-2792.2005
Si T, Luo Y, Bao Z, Zhao H (2015) RNAi-assisted genome evolution in Saccharomyces cerevisiae for complex phenotype engineering. ACS Syn Bio 4:283–291. doi:10.1021/sb500074a
Sun J, Alper H (2015) Metabolic engineering of strains: from industrial-scale to lab-scale chemical production. J Ind Microbiol Biotechnol 42:423–436. doi:10.1007/s10295-014-1539-8
Troutt AB, McHeyzer-Williams MG, Pulendran B, Nossal GJ (1992) Ligation-anchored PCR: a simple amplification technique with single-sided specificity. P Natl Acad Sci USA 89:9823–9825. doi:10.1073/pnas.89.20.9823
van Maris AJ, Winkler AA, Porro D, van Dijken JP, Pronk JT (2004) Homofermentative lactate production cannot sustain anaerobic growth of engineered Saccharomyces cerevisiae: possible consequence of energy-dependent lactate export. Appl Environ Microbiol 70:2898–2905
Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteom MCP 11:492–500. doi:10.1074/mcp.O111.014704
Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. doi:10.1126/science.285.5429.901
Ye L, Kruckeberg AL, Berden JA, van Dam K (1999) Growth and glucose repression are controlled by glucose transport in Saccharomyces cerevisiae cells containing only one glucose transporter. J Bacteriol 181:4673–4675
Ye L, Zhou X, Hudari MS, Li Z, Wu JC (2013) Highly efficient production of l-lactic acid from xylose by newly isolated Bacillus coagulans C106. Bioresour Technol 132:38–44. doi:10.1016/j.biortech.2013.01.011
Zhao K, Qiao Q, Chu D, Gu H, Dao TH, Zhang J, Bao J (2013) Simultaneous saccharification and high titer lactic acid fermentation of corn stover using a newly isolated lactic acid bacterium Pediococcus acidilactici DQ2. Bioresour Technol 135:481–489. doi:10.1016/j.biortech.2012.09.063
Acknowledgments
This work was funded in part by Samsung Electronics Co., Ltd. under the Global Research Outreach Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement/conflict of interest
N. C., J. S., and H. A. do not have any financial or personal relationships that could inappropriately influence or bias the content of the paper. J. L. is an employee of Samsung Corporation.
Additional information
J. J. Lee and N. Crook contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, J.J., Crook, N., Sun, J. et al. Improvement of lactic acid production in Saccharomyces cerevisiae by a deletion of ssb1 . J Ind Microbiol Biotechnol 43, 87–96 (2016). https://doi.org/10.1007/s10295-015-1713-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-015-1713-7