
Abstract
Lactobacilli are widespread in natural environments and are increasingly being investigated as potential health modulators. In this study, we have adapted the broad-host-range vector pNZ8048 to express the mCherry protein (pRCR) to expand the usage of the mCherry protein for analysis of gene expression in Lactobacillus. This vector is also able to replicate in Streptococcus pneumoniae and Escherichia coli. The usage of pRCR as a promoter probe was validated in Lactobacillus acidophilus by characterizing the regulation of lactacin B expression. The results show that the regulation is exerted at the transcriptional level, with lbaB gene expression being specifically induced by co-culture of the L. acidophilus bacteriocin producer and the S. thermophilus STY-31 inducer bacterium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lactobacilli belong to the lactic acid bacteria (LAB) group, and traditionally they have been used for fermented food production. They are widespread in natural environments including niches such as the human and animal gastrointestinal tracts, where they can play important roles as health modulators [2, 23]. Thus, they are increasingly used as functional food ingredients (probiotics), and as vectors for live oral vaccines and drugs [3, 24, 25]. Several Lactobacillus species have been identified to produce bacteriocins, some of them have a dual role, acting as inhibitors at high concentrations and participating in interspecies communication or bacterial crosstalk [8].
Due to the increased interest in lactobacilli health effects, it is relevant to develop genetic tools that can detect regulation in living cells of gene expression such as the production of proteins, including enzymes, from these bacteria [9, 15]. One strategy is the construction of fluorescent proteins to study the effect of lactobacilli in the immune system and their localization within the gastrointestinal tract [2, 6]. Fluorescent proteins could also facilitate the monitoring of these bacteria in biofilm formation and interaction with the host. In addition, fluorescent proteins are used as reporters for transcriptional gene expression and regulation. In our previous work, we designed a mrfp gene codon, optimized for expression in Gram-positive bacteria, from the monomeric variant of the ‘mCherry’ red fluorescent protein (RFP) from Dicosoma sp. [11]. Its use as a reporter in Lactococcus lactis and Enterococcus faecalis was validated by the construction and testing of shuttle vectors based on the pAK80 plasmid [11]. Additional applications of the mCherry-derived vectors in these LAB species have been recently reported [4, 5].
In this study, we have constructed a vector based on the pSH71 replicon [7, 12], to expand the usage of mCherry for analysis of gene expression in Lactobacillus and its usage as a promoter probe validated in L. acidophilus.
Materials and methods
Bacterial strains and culture conditions
Lactobacillus acidophilus CECT 903 from the Colección Española de Cultivos Tipo (Paterna, Spain) and L. acidophilus La5 (Chr. Hansen, Hørsholm, Denmark) strains were grown in MRS broth (Pronadisa, Madrid, Spain) supplemented with 0.05 % l-cysteine hydrochloride (Calbiochem, Merck KGaA, Darmstadt, Germany) and 0.2 % Tween 80 (Oxoid, Hampshire, UK) (MRSCT) at 37 °C. Streptococcus thermophilus STY-31 (Chr. Hansen) was grown in ESTY broth (Pronadisa) supplemented with 0.5 % glucose at 37 °C. Escherichia coli DH-5α [21] was grown in Luria–Bertani broth at 37 °C with vigorous shaking. Streptococcus pneumoniae 708 [18] was grown in AGCH medium [17] supplemented with 0.25 % yeast extract and 0.8 % sucrose at 37 °C without shaking. When necessary, chloramphenicol (Sigma-Aldrich, St. Louis, MO, USA) was added to the culture medium at a final concentration of 10 µg mL−1 for E. coli and 5 µg mL−1 for S. pneumoniae and L. acidophilus. Plate media were prepared by adding agar (Pronadisa) to liquid broth at a final concentration of 1.5 %.
General DNA manipulation and transformation
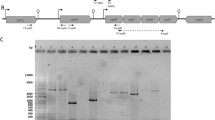
The promoter-probe vector and the expression plasmid constructed in this work were based on the pSH71 replicon [7, 12]. Plasmid pRCR was constructed as follows (Fig. 1). Plasmid pNZ8048 [16] was digested with BglII and SacI to remove the nisin promoter. The resultant 3,168 bp DNA fragment, containing the rolling-circle replicon of the pSH71 plasmid and the chloramphenicol resistance cat gene, was purified from a 0.8 % agarose gel using the QIAquick Gel Extraction Kit (Qiagen Iberia, Madrid, Spain). The mrfp gene encoding the mCherry protein [11] was amplified from plasmid pTVCherry (National Collections of Industrial and Marine Bacteria, Aberdeen, UK) by using the specific primers mCherryF (5′-GGAAGATCTTCCCGAATTCCCCGGGGATCCTCTAGAGGGATACGCACG AGTTTCAACT-3′) and mCherryR (5′-CGCGAGCTCATTTATATAATTCGTCCATGCCACCTGT-3′) to obtain a 801 pb amplicon containing the mrfp gene preceded by the multicloning site BglII, EcoRI SmaI, XmaI BamHI, XbaI. The PCR product was then digested with BglII and SacI restriction enzymes and ligated, with T4 DNA ligase (Thermo Fisher Scientific, Waltham, USA), to the 3,168 bp fragment from pNZ8048. The resulting plasmid, named pRCR (3,960 bp), was established in S. pneumoniae 708 by transformation as previously described [19]. Transformants were selected for chloramphenicol resistance, and the correct nucleotide sequence of the insert, containing a multicloning site and the mrfp gene in pRCR, was confirmed by DNA sequencing at Secugen S.L. (Centro de Investigaciones Biológicas, Madrid, Spain).

Schematic diagram showing the construction of pRCR and pRCR11. For details, see “Materials and methods”. Relevant restriction sites are shown. Specific genes are: mrfp and cat that encode mCherry and the protein responsible for the resistance to chloramphenicol, respectively. P lbaB , promoter of the lactacin B structural gene of Lactobacillus acidophilus La5
The promoter (P lbaB ) of the L. acidophilus La5 lactacin B gene [22] was cloned upstream of mrfp in pRCR, generating the transcriptional fusion P lbaB -mrfp in pRCR11. To this end, an amplicon was generated using the chromosomal DNA of L. acidophilus La5 as template and the primers LBA1797F (5′-AGGAGATCTGCGTACAAAGATGTGGTTAA-3′) and LBA1797R (5′-AGGTCTAGATGAGATTTTTATCTCATTTCAAC-3′) to obtain a 221 pb fragment containing the P lbaB sequence. After digestion with BglII and XbaI, the amplicon was introduced into the multicloning site of pRCR, after digestion with the same restriction enzymes. The resulting plasmid pRCR11 (4,166 bp) was established in E. coli DH5α by transformation as described previously by Hanahan [13]. The presence of the transcriptional fusion in pRCR11 was confirmed by DNA sequencing with primers LBA1797F and LBA1797R.
Plasmid pRCR11 was transferred to L. acidophilus CECT903 cells by electrotransformation as follows. The CECT903 strain was grown under aerobic conditions at 37 °C, without shaking, during 15 h in MRSCT broth supplemented with 1 % glycine. Subsequently, 4 mL of the culture were used to inoculate 200 mL of the same fresh medium, and grown until it reached an OD600 of 0.3–0.4. Cells were collected by centrifugation at 0 °C and 8,600×g for 10 min and washed four times with ice-cold electroporation buffer (HEPES 0.1 mM, sucrose 0.5 M, pH 7.5). Finally, cells were resuspended in 1.6 mL of ice-cold electroporation buffer. 750 µL of cells and 0.5 µg of plasmid DNA were used for electrotransformation. The electroporation conditions were 25 μF, 200 Ω and 2.5 kV in a 0.4-cm cuvette, using a Gene Pulser and a Pulse Controller apparatus (Bio-Rad, Richmond, CA, USA). After electroporation, cells were resuspended in 8 mL of MRSCT broth supplemented with 10 mM CaCl2 and 0.5 M sucrose, incubated aerobically at 37 °C for 3 h, without agitation, and then plated onto MRSCT medium supplemented with 1 % agar and chloramphenicol.
Induction assay and determination of fluorescence
Induction of expression of mCherry from the P lbaB promoter in L. acidophilus CECT903[pRCR11] cells was assayed by co-culturing L. acidophilus CECT903[pRCR11] with the inducer Streptococcus thermophilus STY-31 strain [22]. Co-cultures were carried out in MRSCT medium inoculated with 2 % each of L. acidophilus CECT903 [pRCR11] and S. thermophilus STY-31 overnight cultures.
The levels of fluorescence of the mCherry encoded by pRCR11 and bacterial growth were tested simultaneously with the Varioskan Flash system (Thermo Fisher Scientific, Waltham, MA, USA), which provides quantitative data of cell density via measuring OD at 600 nm and in vivo mCherry expression at an excitation wavelength of 587 nm and an emission wavelength of 612 nm. Background fluorescence of the control strain (L. acidophilus CECT903 [pRCR11] without S. thermophilus STY-31 grown under the same conditions) was used to normalize the fluorescence signals during cultivation. All measurements were performed in sterile 96-well optical bottomed microplates (Nunc, Rochester, NY, USA) with a final assay volume of 300 µL per well by using the microtiter plate assay system Varioskan Flash. The microplates were incubated for 24 h at 37 °C. Measurements were made at 1 h intervals.
Preparation of nucleic acids
For cloning and sequencing experiments, plasmidic DNA was purified from E. coli DH-5α by usage of QIAprep Spin Miniprep and Midiprep kits (Qiagen, Hilden, Germany) or from S. pneumoniae 708 as previously described [21]. For primer extension analysis, a culture of L. acidophilus CECT903[pRCR11] and the co-culture of L. acidophilus CECT903 [pRCR11] and S. thermophilus STY-31 were grown to an OD600 of 1.2 and then used for analysis of mrfp mRNA. Total RNA was isolated with a Ribolyser and Recovery kit from Hybaid (Middlesex, UK) as specified by the supplier. The RNAs were checked for the integrity and yield of the rRNAs by Qubit™ fluorometer (Invitrogen, Madrid, Spain) and by Gel Doc 1000 (Bio-Rad). The patterns of rRNAs were similar in all preparations.
Primer extension analysis
Primer extension analysis was performed by a modification of the method described by Fekete et al. [10]. The start site of lbaB mRNA was detected using the LBABP primer (5′-TGAGTTGAAACTCGGTGCGTATCCTCT-3′) labeled with 6-FAM at its 5′-end (Sigma-Aldrich). Two hundred picomoles of primer were annealed to 40 μg of total RNA. Primer extension reactions were performed by incubation of the annealing mixture with 20 nmol each of dNTP (dATP, dGTP, dCTP and dTTP), 200 U of Maxima Reverse Transcriptase (Thermo Fisher Scientific, Madrid, Spain) in 1× reverse transcriptase buffer (Thermo Fisher) in a final volume of 50 µL at 50 °C for 60 min. Then, the reactions were supplemented with 50 µL of TE (10 mM Tris HCl pH 8.0, 1 mM EDTA) and purified by treatment with phenol (vol:vol) for 5 min at room temperature and ethanol precipitation with three volumes of 100 % ethanol in the presence of 0.3 mM Na acetate. After overnight storage at −20 °C, samples were sedimented by centrifugation at 12,000×g for 30 min at −10 °C and resuspended in TE (12 µL).
Detection and quantification of the reaction products were carried out in a 8 % polyacrylamide gel containing 7 M urea. Bands labeled with 6-FAM were detected and directly quantified with a FujiFilm Fluorescent Image Analyzer FLA-3000 (Fujifilm, Düsseldorf, Germany).
For determination of the length of the extended products, the primer extension reactions were further purified using Agencourt Clean Seq (Beckam Coulter, Alcobendas, Madrid, Spain) and kept frozen at −20 °C until use. Samples were separated on an Abi 3730 DNA Analyzer (Applied Biosystems, Tres Cantos, Madrid, Spain) capillary electrophoresis instrument using techniques and parameters recommended by the manufacturer. A DNA sequence of pRCR11 determined by the dideoxynucleotide method with unlabeled LBABP primer was included in the same capillary in each run to determine fragment length. The Peak Scanner version v1.0 (Applied Biosystems) was used to screen the data and identify major peaks.
Results and discussion
Plasmids containing the mCherry coding gene
Following the construction and analysis of the pTL family of plasmids designed for using mCherry as a reporter in LAB [11], we were unable to transfer any of these plasmids neither to L. casei and L. acidophilus strains nor to S. pneumoniae strains. These pTL plasmids were derived from pAK80, which carries, in addition to the erythromycin resistance marker, two origins of replication, one from the lactococcal plasmid pCT1138 and the other from the E. coli p15A plasmid, and replicates in Gram-positive bacteria by the theta mode mechanism [14]. As an alternative, and with the aim of developing new tools for gene expression analysis in lactobacilli, the use of the replicon of the L. lactis pSH71 plasmid [7, 12] was investigated. This plasmid uses the rolling-circle-type mechanism for replication, and is characterized by a broad host-range, which includes Gram-positive bacteria and E. coli. Therefore, a promoter-probe vector (pRCR) and an expression plasmid (pRCR11) carrying the synthetic mrfp gene optimized for LAB [11] and the chloramphenicol resistance marker were constructed (Fig. 1). pRCR was generated by changing a DNA fragment of pNZ4048 containing the nisA promoter, located between the restriction sites BglII and SacI, by a DNA fragment containing the mrfp gene preceded by a polylinker to facilitate further cloning of transcriptional promoters upstream of the mCherry coding gene. The pRCR plasmid was established in S. pneumoniae 708 by transformation and selection for chloramphenicol resistance. In addition, as predicted, the plasmid was also able to replicate in L. sakei MN1, L. plantarum WCFS1 and E. coli DH5α (personal communications by M. Nacher, A. Pérez-Ramos and M.L. Mohedano, respectively). Consequently, the resulting vector had kept its broad-host-range attribute and had the potential to be used in various LAB species.
To evaluate the functional expression of mCherry in lactobacilli, the region located upstream of the lbaB bacteriocin structural gene from L. acidophilus [22] and carrying the putative promoter P lbaB was cloned upstream of mrfp in pRCR to generate pRCR11. The expression of lactacin B in L. acidophilus has been demonstrated to be inducible by the co-culture with live target bacteria [22]. The mrfp gene was used as reporter to monitor the P lbaB activity during the induction of lactacin B production by the transformation of L. acidophilus CECT 903 with pRCR11. The induction of bacteriocin expression was assayed by co-culturing this strain with S. thermophilus STY-31, a previously identified inducer strain [22]. The functional expression of mCherry under the control of P lbaB and the increase of biomass during cell growth were monitored (Fig. 2). The results revealed that the growth of L. acidophilus CECT 903[pRCR11] was very similar in both single and co-cultures. However, the mCherry activity was detected only in the presence of S. thermophilus STY-31. Moreover, under co-culture conditions the increase of mCherry fluorescence correlated with the growth pattern. These results indicated that up-regulation of lactacin B expression initiates during exponential growth as previously demonstrated [22]. The maximum fluorescence levels were detected at the stationary phase (OD600 = 1.3), consequently growth to this phase was used for further experiments.

Detection of induction of expression of mCherry encoded by pRCR11. Fluorescence (relative fluorescence units; triangles) and growth (OD600; circles) of cultures of L. acidophilus CECT 903[pRCR11] (open symbols) and co-cultures of L. acidophilus CECT 903[pRCR11] and Streptococcus thermophilus STY-31 (closed symbols) grown in MRSCT are depicted. The growth of cultures was monitored at a wavelength of 600 nm. Fluorescence emission of mCherry was recorded at 612 nm after excitation at a wavelength of 587 nm
Transcriptional analysis of the influence of co-culture with S. thermophilus on lactacin B expression in L. acidophilus
Our previous quantitative RT-PCR studies of L. acidophilus La5 lbaB gene expression had shown that in the presence of S. thermophilus STY-31 an increase of the lbaB transcript takes place [22]. Thus, total RNA was extracted from L. acidophilus CECT603[pRCR11] cells grown in the presence or absence of S. thermophilus STY-31 to stationary phase and samples were used for primer extension analysis performed with a 5-end 6-FAM labeled LBABP primer. Analysis of the same volume (6 µL) of both reactions in a polyacrylamide gel detected the extended products encoded by pRCR11 complementary to the lbaB transcript in L. acidophilus CECT603[pRCR11] grown in the presence or absence of S. thermophilus STY-31 (Fig. 3b). In the absence of the inducer two bands with similar intensity were observed, whereas in the co-cultures one more prominent and two minor longer extended products were observed. The fluorescence of the bands was quantified with a fluorescent image analyzer, and the results revealed a fivefold induction due to the presence of S. thermophilus STY-31 (Fig. 3b).

Detection of the start site of the lbaB transcript by primer extension. Reactions containing total RNA isolated from cultures of L. acidophilus CECT 903[pRCR11] alone or in co-culture with S. thermophilus STY-31 were analyzed by capillary electrophoresis in conjunction with DNA sequence of pRCR11 (a) or by 8 % denaturating polyacrylamide gel electrophoresis (b). For primer extension and DNA sequence analysis, primers fluorescently labeled at the 5′-end with 6-FAM, or unlabeled (both having the same DNA sequence) were used, respectively. Extended products ran as if they were, on an average, three nucleotides shorter than the dideoxy sequencing products. The length of the extended products determined by the analysis depicted in a is indicated in the analysis showed in b. The DNA region surrounding the start site of the mRNA is also depicted (c). The start sites of the transcript detected in a are indicated by stars. The −35 and −10 regions of the P lbaB promoter are shown. The inverted repeat, putative binding site of the RR_1798 response regulator, is indicated by arrows
To determine the length of the extended products and the 5-end of the lbaB transcript, the primer extension reactions were also analyzed by capillary electrophoresis in conjunction with a DNA sequence of pRCR11 generated with unlabeled LBABP primer (Fig. 3a). Since we expected low transcript levels of P lbaB in cells grown in mono-culture, we processed 250 nL for the capillary electrophoresis experiments compared to 40 nL derived from cells grown in co-culture. The pattern of the peaks observed (Fig. 3b) correlated with that obtained for the labeled bands in the polyacrylamide gel (Fig. 3a). The two bands detected in cultures of L. acidophilus CECT603[pRCR11] corresponded to extended products of 185 and 187 nt, the first being the major band present in the co-cultures (Fig. 3b). This result located the 5′-end of the lbaB mRNA at a C and A (Fig. 3c), since 6-FAM labeled DNA extended products run as if they were, on an average, three nucleotides shorter than the dideoxy sequencing products [10]. Upstream of the start sites, a putative promoter was detected composed of a −35 (TTGtAa) and a −10 (aATAAT), these sequences being characteristic for the binding of the vegetative σ factor of the bacterial RNA polymerases with an anomalous (too long) spacing of 24 nt (Fig. 3c). Moreover, the two start sites for transcription are included in one of the arms of the inverted repeat characteristic for binding of transcriptional regulators. This location predicts that the binding of a protein to the inverted repeat will impair initiation of transcription catalyzed by the RNA polymerase. The expression of the lactacin B operon is regulated by the response regulator RR_1798 which is part of a three-component regulatory system composed of the inducing peptide IP_1800, the HK_1799 histidine kinase and the RR_1798 response regulator [1, 22]. Thus, it seems that under uninduced conditions competition between the RNA polymerase and RR_1798 for binding to the upstream region of lbaB gene will result in low levels of the transcript starting at the two nucleotides G and C. Then, in the presence of bacteria that compete for the environmental niche, HK_1799 would sense its presence and, by modification of the RR_1798, would impair the repression of transcription of lbaB and result in an increase of lactacin B levels. We have previously demonstrated that the production of lactacin B by L. acidophilus is controlled by an autoinduction mechanism involving a secreted peptide and by co-culture with live inducer cells [22]. These characteristics of induction of bacteriocin production through autoinduction and co-culture have been recently described to be widespread among bacteriocinogenic L. plantarum strains [20]. The use of mCherry as a promoter probe in pRCR11 has allowed us to locate the region where the lbaB transcriptional regulation is specifically induced by co-culture of the lactacin B producer with the inducing bacteria.
In conclusion, the rolling-circle-type mechanism for replication of pRCR has broadened the host-range in LAB of the mCherry-based vectors pTLR. Indeed, the promoter-probe vector pRCR has demonstrated to be suitable for characterization of complex promoter induction mechanisms such as those related to bacteriocin production by L. acidophilus.
References
Altermann E, Russell WM, Azcarate-Peril MA, Barrangou R, Buck BL, McAuliffe O, Souther N, Dobson A, Duong T, Callanan M, Lick S, Hamrick A, Cano R, Klaenhammer TR (2004) Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc Natl Acad Sci USA 102:3906–3912
Bao S, Zhu L, Zhuang Q, Wang L, Xu PX, Itoh K, Holzman IR, Lin J (2013) Distribution dynamics of recombinant Lactobacillus in the gastrointestinal tract of neonatal rats. PLoS One 8:e60007
Bull M, Plummer S, Marchesi J, Mahenthiralingam E (2013) The life history of Lactobacillus acidophilus as a probiotic, a tale of revisionary taxonomy, misidentification and commercial success. FEMS Microbiol Lett 349:77–87
Campelo AB, Roces C, Mohedano ML, López P, Rodríguez A, Martínez B (2014) A bacteriocin gene cluster able to enhance plasmid maintenance in Lactococcus lactis. Microb Cell Fact 13:77
Cebrián R, Rodríguez-Ruano S, Martínez-Bueno M, Valdivia E, Maqueda M, Montalbán-López M (2014) Analysis of the promoters involved in enterocin AS-48 expression. PLoS One 9:e90603
Chen Z, Lin J, Ma C, Zhao S, She Q, Liang Y (2014) Characterization of pMC11, a plasmid with dual origins of replication isolated from Lactobacillus casei MCJ and construction of shuttle vector with each replicon. Appl Microbiol Biotechnol 98:5977–5989
De Vos WM (1987) Gene cloning and expression in lactic streptococci. FEMS Microbiol Rev 46:281–295
Dobson A, Cotter PD, Ross RP, Hill C (2012) Bacteriocin production, a probiotic trait? Appl Environ Microbiol 78:1–6
Duong T, Miller MJ, Barrangou R, Azcarate-Peril MA, Klaenhammer TR (2010) Construction of vectors for inducible and constitutive gene expression in Lactobacillus. Microb Biotech 4:357–367
Fekete RA, Miller MJ, Chattoraj DK (2003) Fluorescently labeled oligonucleotide extension, a rapid and quantitative protocol for primer extension. Biotechniques 35:90–98
García-Cayuela T, Gómez de Cadiñanos LP, Mohedano ML, Fernández de Palencia P, Boden D, Wells J, Peláez C, López P, Requena T (2012) Fluorescent protein vectors for promoter analysis in lactic acid bacteria and Escherichia coli. Appl Microbiol Biotechnol 96:171–181
Gasson MJ (1983) Plasmid complements of Streptococcus lactis and other lactic streptococci after protoplast-induced curing. J Bacteriol 154:1–9
Hanahan D (1985) Techniques for transformation of E. coli. In: Glover DM (ed) DNA cloning, a practical approach, vol 1. IRL, Oxford, pp 109–135
Israelsen H, Madsen SM, Vrang A, Hansen EB, Johansen E (1995) Cloning and partial characterization of regulated promoters from Lactococcus lactis Tn917-lacZ integrants with the new promoter probe vector, pAK80. Appl Environ Microbiol 61:2540–2547
Kleerebezem M, Beerthuyzen MM, Vaughan EE, De Vos WM, Kuipers O (1997) Controlled gene expression systems for lactic acid bacteria, transferable nisin-inducible expression cassettes for Lactococcus, Leuconostoc, and Lactobacillus spp. Appl Environ Microbiol 63:4581–4584
Kuipers OP, De Rayter PG, Kleerebezem M, De Vos WM (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64:15–21
Lacks S (1968) Genetic regulation of maltosaccharide utilization in Pneumococcus. Genetics 60:685–706
Lacks SA, Greenberg B (2001) Constitutive competence for genetic transformation in Streptococcus pneumoniae caused by mutation of a transmembrane histidine kinase. Mol Microbiol 42:1035–1045
Lacks SA, López P, Greenberg B, Espinosa M (1986) Identification and analysis of genes for tetracycline resistance and replication functions in the broadhost-range plasmid pLS1. J Mol Biol 192:753–765
Maldonado-Barragán A, Caballero-Guerrero B, Lucena-Padrós H, Ruiz-Barba JL (2013) Induction of bacteriocin production by coculture is widespread among plantaricin-producing Lactobacillus plantarum strains with different regulatory operons. Food Microbiol 33:40–47
Mohedano ML, Overweg K, De la Fuente A, Reuter M, Altabe S, Mulholland F, De Mendoza D, López P, Wells J (2005) Inducible expression of the essential response regulator YycF in Streptococcus pneumoniae modulates expression of genes involved in fatty acid biosynthesis and affects membrane composition. J Bacteriol 178:2357–2367
Sambrook J, Russell DW (2001) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Tabasco R, García-Cayuela T, Peláez C, Requena T (2009) Lactobacillus acidophilus La-5 increases lactacin B production when it senses live target bacteria. Int J Food Microbiol 132:109–116
Van Baarlen P, Wells JM, Kleerebezem M (2013) Regulation of intestinal homeostasis and immunity with probiotic lactobacilli. Trends Immunol 34:208–215
Wang A, Yu H, Gao X, Li X, Qiao S (2009) Influence of Lactobacillus fermentum I5007 on the intestinal and systemic immune responses of healthy and E. coli challenged piglets. Antonie Van Leeuwenhoek 96:89–98
Wells JM (2011) Mucosal vaccination and therapy with genetically modified lactic acid bacteria. Ann Rev Food Sci Technol 2:423–445
Acknowledgments
This study was supported by Grants AGL2012-40084-C03-01, AGL2012-35814 and RM2011-00003-00-00 from the Spanish Ministry of Economics and Competitiveness and by European Commission FP7 Initial Training Network (contract 238490). We thank Dr. Stephen Elson for the critical reading of the manuscript. We also thank M. Angeles Corrales for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
M.L. Mohedano and T. García-Cayuela contributed equally to the work.
Rights and permissions
About this article

Cite this article
Mohedano, M.L., García-Cayuela, T., Pérez-Ramos, A. et al. Construction and validation of a mCherry protein vector for promoter analysis in Lactobacillus acidophilus . J Ind Microbiol Biotechnol 42, 247–253 (2015). https://doi.org/10.1007/s10295-014-1567-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1567-4