
Abstract
Sugarcane ethanol production occurs in non-sterile conditions, and microbial contamination can decrease productivity. In this study, we assessed the microbial diversity of contaminants of ethanol production in an industrial facility in Brazil. Samples obtained at different stages were analyzed by pyrosequencing-based profiling of bacterial and archaeal 16S rRNA genes and the fungal internal transcribed spacer region. A total of 355 bacterial groups, 22 archaeal groups, and 203 fungal groups were identified, and community changes were related to temperature changes at certain stages. After fermentation, Lactobacillus and unclassified Lactobacillaceae accounted for nearly 100 % of the bacterial sequences. Predominant Fungi groups were “unclassified Fungi,” Meyerozyma, and Candida. The predominant Archaea group was unclassified Thaumarchaeota. This is the first work to assess the diversity of Bacteria, and Archaea and Fungi associated with the industrial process of sugarcane-ethanol production using culture-independent techniques.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brazil is the world’s largest producer of sugarcane [82] and the second largest producer of sugarcane ethanol [69]. A total of 8.81 million hectares of sugarcane were harvested in Brazil during the 2013–2014 season, most of it in the state of São Paulo (4.56 million hectares). Productivity in the 2013–2014 season was 74.8 tons per hectare, an increase of 7.9 % compared with the previous harvest of 69.4 tons per hectare [26]. The income generated by ethanol exports was approximately $1.87 billion in 2013 [14].
There are currently 390 active sugar- and ethanol-producing plants in Brazil [15]. Many of these plants operate under the biorefinery concept, producing not only fuel ethanol and sugar but also products such molasses, bagasse, vinasse, filter cake, energy (i.e., by burning bagasse), and yeast to be used as a protein-rich component of animal feed. The ability to produce multiple products from sugarcane gives the ethanol industry flexibility and contributes to its sustainability by increasing the economic value of the process as a whole [11]. Brazil plays an important role in this sector, not only because of its large ethanol and sugarcane production, but also because of research that has led to the development of new higher-yielding sugarcane varieties and the optimization and mass adoption of flex-fuel engines in Brazil [11]. Because sugarcane is grown in the vicinity of industrial plants, Brazil also has unparalleled logistics and low sugarcane shipping costs [25].
Ethanol can be produced directly from sugarcane juice or from a mixture of sugarcane juice and molasses. In the first steps of the process, sugarcane juice is decanted, heated, and concentrated to produce the must, which is full of sugars and ready for fermentation. Saccharomyces cerevisiae yeasts are then added to the must, and fermentation is run for 8–12 h, producing a fermented must (wine) containing 7–10 % ethanol. The yeasts are then recycled, and the wine is distilled to recover the ethanol [11].
Although ethanol production from sugarcane is well-established, improvements are always needed. Industrial ethanol production process does not occur in sterile conditions; microbial contamination is expected and tolerated [76]. However, contaminants can decrease productivity by competing for nutrients needed for yeast growth and fermentation and by producing organic acids that inhibit yeast metabolism [5]. High levels of bacterial byproducts (e.g., acetic and lactic acid) can lead to costly downtime spent cleaning the machinery [10, 75]. Other problems caused by contaminant microorganisms are gum production, yeast flocculation, and synthesis of toxins that inhibit yeast and decrease their viability, all of which can reduce productivity [62].
Classic microbiology techniques have been used to isolate and characterize contaminants [20, 21, 35, 54], which include Gram-negative and Gram-positive bacteria of the genera Pediococcus, Enterococcus, Acetobacter, Gluconobacter, and Clostridium [10, 76]. However, the most common species belong to Lactobacillus, a genus of fast-growing bacteria that tolerates ethanol and low pH [6, 10, 76]. In addition, other types of yeast, the most important of which is Dekkera bruxellensis, can also be contaminants of the process and are even more difficult to control without directly affecting S. cerevisiae [7].
High-throughput culture-independent methods have been used to describe microbial communities in different industrial processes [28, 38, 59, 83, 87] but have not yet been used to describe microbial contaminants in ethanol production. This is the first study using pyrosequencing to characterize the microbial contaminants present in different steps of the industrial sugarcane ethanol production process.
Materials and methods
Sampling
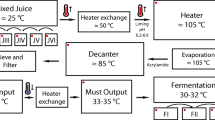
Samples were obtained in July 2012 at a distillery in the state of Goiás that produces more than 100 million liters of ethanol per season. Triplicate samples (500 mL each) of the following stages of the ethanol production process were collected in sterilized glass bottles: sugarcane juice, mixed juice, clarified juice, evaporated juice, must, and wine (Fig. 1). The temperature and pH of the samples were measured on site with a thermometer and pH-Fix test strips (Macherey-Nagel GmbH & Co, Düren, Germany), respectively. Samples were then transported to the lab on ice and stored at −80 °C until used for DNA extraction.

Schematic of the sugarcane ethanol production process. Obtained samples are indicated in parenthesis
Total DNA extraction
Aliquots of each sample (approximately 40 mL) were centrifuged at 18,650g for 30 min, and DNA was extracted from the pellet with FastDNA® Spin Kit for Soil (MP Biomedicals, LLC, Santa Ana, CA, USA), according to the manufacturer’s instructions.
Total DNA was used as template for polymerase chain reaction (PCR) amplification of bacterial and archaeal 16S rRNA genes and the fungal internal transcribed spacer (ITS) region. The primers used were 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) [49] and 519R (5′-GWA TTA CCG CGG CKG CTG-3′) [81] for bacteria; 109F (5′-ACK GCT CAG TAA CAC GT-3′) [85] and 915R (5′-GTG CTC CCC CGC CAA TTC CT-3′) [77] for archaea; and ITS1F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) [36] and ITS4 (5′-TCC TCC GCT TAT TGA TAT GC-3′) [84] for fungi. Adapters used as priming sites for both amplification and sequencing (454 Life Sciences, Branford, CT, USA) were ligated to the 5′ end of the primer sequences (adapter A for forward primers, and adapter B for reverse primers). The conditions for PCR amplification with the 27F/519R primer pair were: denaturation at 95 °C for 3 min followed by 25 cycles of denaturing at 94 °C for 30 s, annealing at 52 °C for 30 s, and extension at 72 °C for 1 min 40 s, with a final extension step at 72 °C for 7 min. The conditions for PCR amplification with the 109F/915R primer pair were: denaturation at 94 °C for 5 min followed by 25 cycles of denaturation at 94 °C for 1 min, annealing at 52 °C for 1 min, extension at 72 °C for 1 min 30 s, with a final cycle of 52 °C for 1 min and 72 °C for 6 min. The conditions for PCR amplification with the ITS1F/ITS4 primer pair were: denaturation at 95 °C for 5 min followed by 30 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min, with a final extension cycle at 72 °C for 10 min. Each 20-μL PCR reaction contained 0.25 μM each primer, 0.25 mM each dNTP, 0.4 U Taq DNA polymerase, 1× reaction buffer (with 1.5 mM MgCl2), and approximately, 10 ng total DNA. Amplifications were performed using an Applied Biosystems GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA). Ten reactions were performed for each sample, and the PCR products were pooled and purified with the GeneJET PCR Purification Kit (Thermo Fisher Scientific, Waltham, MA, USA). After purification, the PCR products were quantified using the Qubit® fluorometer (Invitrogen, Carlsbad, CA, USA) and NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific). Pyrosequencing of the purified PCR products was performed on one-fourth of a sequencing plate using GS FLX Titanium platform at Macrogen, South Korea.
Pyrosequencing analysis of microbial communities
Sequences were analyzed using the software package Mothur [74]. Denoising of the raw data was performed with the tool shhh.flows, an adaptation of the PyroNoise algorithm [67]. Multiplex identifier barcodes and adapters were removed, as well as sequences shorter than 250 bp. Bacteria and Archaea sequences were aligned using the align.seqs tool against the SILVA databases [65]. Fungi sequences were aligned using the online alignment tool Multiple Alignment using Fast Fourier Transform (MAFFT). Chimeras were removed with the tools chimera.uchime (Bacteria and Archaea) and chimera.perseus (Fungi). Phylogenetic classification of the microorganisms was performed using the SILVA databases [65] for Bacteria and Archaea and the UNITE database [47] for Fungi. The number of sequences in each sample was normalized to the smallest number of sequences for each group (Bacteria, Archaea, and Fungi). Sequences were clustered into operational taxonomic units (OTUs) using the average-neighbor method at a 3 % distance threshold. Mothur was used to calculate diversity and richness indices and to carry out molecular variance analysis (AMOVA). Principal component analysis (PCA) was carried out using the UniFrac algorithm to perform qualitative and quantitative comparisons among bacterial communities [53]. The level of significance was set at 0.05.
Results
The temperatures of samples obtained during different stages of ethanol production varied from 30 °C (sugarcane juice) to 92 °C (evaporated juice), whereas the pH of samples was approximately constant throughout the process (Table 1).
Pyrosequencing of bacterial and archaeal 16S rRNA genes and the fungal ITS region generated a total of 172,450 raw sequences. After quality control and removal of chimeric sequences, 140,791 sequences longer than 250 bp remained for subsequent analyses. Between 1,267 and 10,313 high-quality sequence reads were obtained per sample (Table 2).
Bacterial diversity
After normalization, 1,267 sequences for each sample were analyzed for the domain Bacteria, which identified 21 known phyla or candidate divisions in the six stages of ethanol production. Four phyla (Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes) were predominant in the samples, as well as unclassified Bacteria (Fig. 2a). The phylum Firmicutes was dominant in sugarcane juice, clarified juice, evaporated juice, and wine samples, accounting for 49.5–98.2 % of the bacterial sequences, whereas Proteobacteria was dominant in mixed juice and must samples, accounting for 38.0–91.1 % of the sequences.

Most abundant bacterial a phyla, b classes and c genera in different stages of sugarcane ethanol production
Within the phylum Firmicutes, the most abundant class was Bacilli (5.5–98.2 % of the sequences), which was predominant in sugarcane juice, mixed juice, clarified juice, evaporated juice, and wine (Fig. 2b). Gammaproteobacteria was the most abundant class within the phylum Proteobacteria (0.03–87.4 % of the sequences) and the predominant class in must. Unclassified Bacteria were more common in mixed juice than other stages of ethanol production (5.0 % of the sequences).
At the genus level, a total of 355 groups were identified, 215 of which were known genera, and 140 were unclassified groups (Fig. 2c). Leuconostoc was the most abundant genus in sugarcane juice (49.9 % of the sequences), Lactobacillus was predominant in mixed juice and wine samples (16.7–62.6 % of the sequences), Tatumella was predominant in clarified juice and must samples (11.1–77.7 % of the sequences), and Paenibacillus was predominant in evaporated juice (41.4 % of the sequences).
The sugarcane ethanol production stage with the highest number of bacterial OTUs, defined at a 3 % dissimilarity level, was mixed juice (385 OTUs), which showed higher than expected richness and diversity according to the Chao1, abundance-based coverage estimator (ACE), Shannon, and inverse Simpson indices (Table 2). This sample also had the lowest Good’s coverage value. Wine samples had the smallest number of OTUs (19 OTUs) and the lowest expected richness. Must samples had the lowest diversity estimate.
Good´s coverage estimator showed that the number of sequences obtained for evaporated juice, must, and wine were able to cover the existing bacterial diversity at these stages (Table 2). On the other hand, samples of sugarcane juice, clarified juice and mixed juice did not cover the total diversity. Similarly, results of Unifrac PCA, both weighted and unweighted, indicated that the bacterial communities present in evaporated juice and must samples did not differ significantly (Fig. 3). However, the other stages of sugarcane ethanol production, the bacterial communities differed (AMOVA, p < 0.001).

Principal component analysis (PCA) of sequences of the domain Bacteria based on a weighted UniFrac (quantitative analysis) and b unweighted UniFrac (qualitative analysis)
Archaeal diversity
Pyrosequencing of the archaeal 16S rRNA genes was possible only for the sugarcane juice and mixed juice samples. The number of sequences was normalized to 3,130 for these ethanol production stages (Table 2). Only two phyla, Euryarchaeota and Thaumarchaeota, were detected in these samples, along with unclassified Archaea (Fig. 4a). Thaumarchaeota was the main phyla detected in both stages, accounting for 90.9 % of the archaeal sequences in sugarcane juice and 96.5 % of the sequences in mixed juice, whereas Euryarchaeota accounted for only 8.6 % of the sequences in sugarcane juice and 3.1 % of the sequences in mixed juice, and unclassified Archaea sequences accounted for 0.5 % of the sequences in sugarcane juice and 0.4 % of the sequences in mixed juice.

Most abundant archaeal a phyla, b classes and c genera in sugarcane juice and mixed juice
At the class level, the most abundant group was unclassified Thaumarchaeota (48.0 % of the sequences in sugarcane juice and 58.5 % of the sequences in mixed juice), followed by ArcC-u-cD06 (22.1 % of the sequences in sugarcane juice and 23.3 % of the sequences in mixed juice) (Fig. 4b). A few representatives of South African Gold Mine Gp 1, Terrestrial Group, Marine Group I, and Miscellaneous Crenarchaeotic Group were detected in mixed juice only (data not shown).
At the genus level, 22 groups were found in both samples, four of which were known genera, three were candidate genera, and 15 were unclassified genera (Fig. 4c). The most predominant group was unclassified Thaumarchaeota (48.0 % of the sequences in sugarcane juice and 58.5 % of the sequences in mixed juice), followed by unclassified ArcC-u-cD06 (22.1 % of the sequences in sugarcane juice and 23.3 % of the sequences in mixed juice) and unclassified Soil Crenarchaeotic Group (14.5 % of the sequences in sugarcane juice and 6.4 % of the sequences in mixed juice) (Fig. 4c). As shown in Table 2, more OTUs were detected in mixed juice samples (98 OTUs) than in sugarcane juice samples (88 OTUs). Species richness was higher in mixed juice, as assessed by abundance-based estimators (Chao1 and ACE) and the inverse Simpson’s diversity index. However, the Shannon diversity index value was slightly higher for sugarcane juice. The Good´s value for estimated coverage was 98–99 % (AMOVA, p = 0.086) (Table 2).
Fungal diversity
Analysis of fungal diversity by pyrosequencing of the ITS region was performed only for sugarcane juice and mixed juice samples. Three phyla (Ascomycota, Basidiomycota, and Chytridiomycota), as well as unclassified Fungi, were identified in these samples. Ascomycota was the predominant phylum, accounting for 60.3–64.4 % of the sequences, followed by unclassified Fungi, accounting for 30.0–31.9 % of the sequences (Fig. 5a). Phylum Chytridiomycota was represented by just one sequence in the sugarcane juice sample.

Most abundant fungal a phyla, b classes and c genera in sugarcane juice and mixed juice
The most abundant classes in both samples belonged to the phylum Ascomycota (Fig. 5b). Saccharomycetes were predominant in sugarcane juice, accounting for 27.0 % of the sequences, whereas Sordariomycetes was predominant in mixed juice, accounting for 19.8 % of the sequences. The class Eurotiomycetes was more abundant in mixed juice samples (18.4 % of the sequences) than in sugarcane juice samples (9.7 % of the sequences), whereas Lecanoromycetes and Exobasidiomycetes were found only in sugarcane juice (0.01 % of the sequences for both classes) (data not shown). More representatives of the class Agaricomycetes were found in sugarcane juice (2.2 % of the sequences) than in mixed juice (0.5 % of the sequences).
At the genus level, 203 groups were identified, including 59 unclassified groups and 144 known genera (Fig. 5c). Unclassified Fungi was the most abundant group in both samples. Unclassified Sordariomycetes and unclassified Trichocomaceae were more abundant in mixed juice (7.3 and 10.7 % of the sequences, respectively) than in sugarcane juice (3.5 and 5.8 % of the sequences, respectively), and Candida was more abundant in sugarcane juice (8.7 % of the sequences) than in mixed juice (5.72 % of the sequences).
The number of OTUs, defined at a 3 % dissimilarity level, was similar for both samples (495–496 OTUs), as were values for richness and diversity estimators (Table 2). Good´s value estimated coverage at 96 % (AMOVA, p = 0.013) (Table 2).
Discussion
This is the first study to describe microbial communities associated with industrial sugarcane ethanol production using culture-independent methods. Despite advances in current molecular techniques, most studies in this area have used classic cultivation techniques [50, 54, 70], which does not provide a complete picture of microbial diversity. Further, most studies focus on contaminants found in the must, wine, and yeast cream [18, 30, 31, 54]. Contaminants can inhibit yeast growth, decrease carbohydrate utilization, increase acidity, and reduce ethanol production by as much as 22 % [57, 78]. Therefore, we performed a thorough analysis of the bacterial, archaeal, and fungal communities present in different stages of the industrial ethanol production process, including the early steps that are usually neglected.
In a typical plant, such as the one that was the focus of this study, sugarcane is washed and chopped after its arrival, and the juice is then extracted by mills, with a sugar extraction efficiency reaching 96 % [24]. The remaining bagasse is sent to boilers to be burned as fuel, often producing enough energy to make the plants self-sufficient and frequently generating a surplus. Larger impurities are removed from the extracted juice with fixed or vibrating screens, and the pH is adjusted to 6.8–7.2 with calcium hydroxide. The juice is heated to 105 °C, and heavier particles are decanted to produce the clarified juice. Sugars are concentrated by evaporation of the clarified juice at 115 °C. The evaporated juice is then cooled to approximately 30 °C. The sugar concentration is adjusted to produce the must, which is inoculated with yeasts for 8–12 h of fermentation. Yeasts are removed by centrifugation, and the resulting wine is distilled to recover the ethanol [11, 24, 37] (Fig. 1). In this work, for a comprehensive analysis of the microbial community present in the ethanol production process, samples were obtained at six different stages going from the early to late stages of the process (sugarcane juice, mixed juice, clarified juice, evaporated juice, must, wine).
For sugarcane juice, previous studies have focused on endophytic diazotrophic bacteria such as Gluconacetobacter diazotrophicus [63], because of its ability to fix nitrogen and promote plant growth, as well as bacteria of the genera Herbaspirillum and Burkholderia [12, 34, 66]. Other genera of endophytic bacteria that have been isolated and studied include Enterobacter, Erwinia, Klebsiella [68], Pantoea [52], and Bacillus [68]. In this study, the most abundant genus in sugarcane juice was Leuconostoc (Fig. 2c) which belongs to the phylum Firmicutes. This genus was also predominant in mixed juice and clarified juice. Leuconostoc are Gram-positive, non-spore–forming immobile cocci [33, 71] that can be found in environments associated with plants and decaying plant material [71]. In mixed juice, the predominant genus was Lactobacillus (Fig. 2c), also a Firmicutes, which is naturally present in plants, soil, and the gastrointestinal, urogenital tracts, oral cavity, and skin of animals [55]. Thus, our data show that the microbes present in sugarcane juice and mixed juice include both endophytic and epiphytic bacteria, along with other microorganisms naturally present in soil. Diversity indices and Good´s coverage value (Table 2) suggest a higher diversity for the mixed juice stage, likely because the sugarcane juice sample was derived from a single variety of sugarcane, whereas the mixed juice sample was obtained from the mill, where sugarcane of different varieties and from different farms had been crushed. In addition, feedstock and soil are not the only sources of microbial contaminants. Some of the contaminants are likely associated with the chopping and milling equipment.
To date, few studies have characterized sugarcane-associated fungal microbiota. In sugarcane cultivars from Iraq, Abdullah and Saleh [1] identified, through cultivation, Ascomycetes of the following genera: Arxiomyces, Chaetomium, Coniochaeta, Kerinia, and Leptosphaeria. In another study, the same authors [2] reported 16 mitosporic fungi of the genera Alternaria, Bipolaris, Curvularia, Exserohilum, and Drechslera. Azeredo et al. [29] isolated yeasts from the leaves, stems, and rhizosphere of sugarcane at different stages of development and reported that the predominant species were Cryptococcus laurentii, Cryptococcus albidus, Rhodotorula mucilaginosa, and Debaryomyces hansenii. This study reported that Pichia, Torulaspora, Tremella, and Saccharomyces are also associated with sugarcane leaves, stems, and rhizosphere [29].
In our study, pyrosequencing analysis of the fungal ITS region revealed the presence of 144 known genera and 59 groups of unclassified Fungi in the sugarcane juice and mixed juice stages of sugarcane ethanol production. Although amplification products were also obtained for wine samples, pyrosequencing was unsuccessful, probably due to technical issues. For the other steps of ethanol production, amplification of fungal sequences was not possible even after repeated attempts. This may have been due to the small amounts of fungal DNA present in the samples or primers that were not adequate to cover the fungal diversity present. Most of the fungal sequences that were amplified belonged to the phylum Ascomycota, followed by unclassified Fungi and the phylum Basidiomycota. In all samples, only one sequence belonging to the phylum Chytridiomycota was identified (Fig. 5a). At the genus level, most of the sequences detected in sugarcane juice and mixed juice could not be classified, suggesting a high but largely unknown fungal diversity, despite many previous studies using culture-dependent methods. Similar to the results of previous studies [4, 17, 51], Candida and Meyerozyma were among the predominant genera (Fig. 5c). Studying acute contamination episodes in distilleries from the Brazilian states of Pernambuco and Paraiba, Basílio [4] identified D. bruxellensis, Candida tropicalis, Pichia galeiformis, and Candida as the main yeasts present. However, D. bruxellensis, considered to be the most damaging species of contaminant yeasts [7], was not detected in our samples. Cabrini and Gallo [17] found that the predominant contaminant yeast genus in the Pedra ethanol plant in Brazil was Saccharomyces, but other genera such as Candida, Torulopsis, Pichia and Schizosaccharomyces were also present.
Differences between the fungal groups found in sugarcane juice and mixed juice were also observed, suggesting that microorganisms are brought into the ethanol production process with feedstock and soil impurities. Results from AMOVA suggest that the fungal communities differ significantly between stages (p = 0.013). Good´s value estimated coverage was 96 % (AMOVA, p = 0.013) (Table 2), indicating that the number of sequences analyzed were sufficient to cover the expected diversity in these stages.
Archaeal diversity has been studied in various environments such as oceans [32, 46, 72], rumen [27, 86], and soil [9, 19, 40]. However, to date no studies have described the diversity of Archaea in sugarcane or in the sugarcane ethanol production process. Although we were able to amplify archaeal 16S rRNA genes from the sugarcane and mixed juice samples, amplification of archaeal sequences was not successful for the other ethanol production stages despite repeated attempts. The possibility that archaea were absent in these samples cannot be ruled out. However, it is also possible that archaea are present in very low numbers, making PCR amplification difficult, or that the primers used were not adequate to cover the archaeal diversity present. Another possibility is that archaea in the first steps of the process are originally present in association with the plant, and in further stages, they are not able to compete with the other microorganisms present.
The predominant archaeal phylum in both sugarcane juice and mixed juice samples was Thaumarchaeota, followed by Euryarchaeota (Fig. 4a). The phylum Thaumarchaeota is one of the most abundant archaeal groups on Earth, and it comprises a range of archaea from different environments [16, 64] including ammonia-oxidizing microorganisms and archaea with unknown energy metabolism. Members of the phylum Euryarchaeota are present in many environments such as the rumen of goats [27], hydrothermal vents [58], solfataric fields [73], soil [42], rice roots [39], and other places [42]. Although not completely characterized in terms of metabolism, they include known psychrophiles, thermophiles, mesophiles, halophiles, and alkaliphiles. At the genus level, sugarcane juice and mixed juice were dominated by unclassified sequences such as unclassified Thaumarchaeota. Only one candidate genus was among the most predominant groups in sugarcane juice and mixed juice, Candidatus Nitrososphaera (Fig. 4c). The provisional “Candidatus” status is given to cultured prokaryotes whose characterization needed for the International Code of Nomenclature of Bacteria description is incomplete [60].
Results of AMOVA showed that archaeal communities did not differ significantly between stages of ethanol production (p = 0.086). Good´s coverage estimator (Table 2) indicates a high coverage of Archaea diversity.
For stages of ethanol production following the sugarcane juice and mixed juice stages, only bacterial diversity could be analyzed. The production of clarified juice requires correcting the pH of mixed juice to 6.8–7.2 and increasing the temperature to 105 °C in an attempt to eliminate contaminating microorganisms (Fig. 1), accounting for the observed shift in predominant bacterial populations in the subsequent stages of ethanol production (i.e., clarified juice, evaporated juice, must, and wine). The predominant bacterial genus in clarified juice was Tatumella, an Enterobacteriaceae that has been isolated from fruits, soil, and human samples [13] (Fig. 2c). Good´s coverage estimator (Table 2) indicates that the number of sequences analyzed was insufficient to cover the existing bacterial diversity at that stage. However, it is clear that the higher temperature of 105 °C (83 °C at the time it was measured) acted as a selective pressure to decrease bacterial diversity, because the Good´s coverage for clarified juice is higher than that for mixed juice.
Sugars are concentrated in the evaporated juice stage, and the temperature reaches 115 °C (Fig. 1). Good´s coverage estimator indicates that the sequencing effort was sufficient to estimate bacterial diversity in this stage of ethanol production (Table 2). The second rise in temperature occurred in this stage, and this temperature (115 °C) was the highest in the process. Few bacteria are likely to survive this high temperature and the osmotic pressure due to the high concentrated sugars, resulting in lower bacterial diversity. The predominant genus in this stage was Paenibacillus, Gram-positive, spore-forming rod bacteria [41, 79] (Fig. 2c). Spore formation would clearly give this group a selective advantage under the high temperature conditions of this stage, as bacterial spores are resistant to processes that would normally kill vegetative cells such as heating, freezing, dehydration, and radiation [80]. In addition, high sugar concentrations increase the thermal resistance of some bacteria and their spores [8]. Furthermore, the genera predominant in evaporated juice (Paenibacillus) and clarified juice (Tatumella) produce biofilms, which can be resistant to high temperature, cleaning products, ethanol, and acids [48, 79]. This ability can help explain the presence of these genera in stages of sugarcane ethanol production where the temperature is high enough to eliminate most bacteria.
Must is essentially a cooled (45 °C) concentrated sugar solution that is ready to receive yeasts for fermentation (Fig. 1). Gallo [35] studied the microbiota of must before fermentation, reporting that the predominant genera were the Gram-positive rods Bacillus and Lactobacillus, with Enterobacteriaceae accounting for only 9.52 % of the isolated microorganisms. In our study, the predominant genus detected in must (before fermentation) was Tatumella, a member of the Enterobacteriaceae family. This genus, like many Enterobacteriaceae, is capable of fermenting glucose and sucrose, suggesting that the amount of sugar in the must favored its growth [45] (Fig. 2c). There is also the possibility that the equipment was already contaminated by bacteria from previous fermentations, further favoring the presence of Tatumella and unclassified Enterobacteriaceae in this stage of the process because of their potential ability to form biofilms. The value for Good´s coverage estimator was high (97 %), indicating that must bacterial diversity was adequately covered by the sequencing effort (Table 2).
In this study, the greatest change in the bacterial community occurred in wine stage, which was analyzed after yeast removal by centrifugation. Lactobacillus was the predominant genus, followed by unclassified Lactobacillaceae; these groups together accounted for 97.7 % of the sequences (Fig. 2c). The Good´s coverage value was 1.00 (Table 2), indicating complete coverage of bacterial diversity. Bacteria of the genera Bacillus and Lactobacillus have been reported to be major contaminants of ethanol production [3, 54, 76]. Lactic acid bacteria not only cause problems during fermentation in ethanol production from sugarcane, but also in ethanol production from corn [76], wheat [43], tapioca, barley [21, 22], malt [56], triticale, and rye [50]. As observed in this work, these bacteria may originate from earlier steps of the production process or from feedstock [48] and may be the most problematic contaminants, because they are fast-growing, resistant to high temperature and low pH, and tolerant to ethanol, rapidly outnumbering fermenting yeasts [61].
Because of the high number of Lactobacillus sequences in the wine stage compared with earlier stages in the process, it is possible that enrichment for Lactobacillus occurred throughout the process. Alternatively, their source may be equipment contamination from previous fermentations. This genus is also capable of producing biofilms [44, 48], indicating the importance of testing equipment for bacterial contamination. Efficient cleaning of the equipment may be an important measure to control contaminants.
In the course of ethanol production, contaminants such as Lactobacillus can obstruct pipes, sieves, centrifuges, heat sinks, increase flocculation of yeasts and decrease fermentation activity. Flocculation and organic acids produced by bacteria can also decrease yeast viability, reducing ethanol production, leading to stuck fermentations and causing shutdown of facilities for cleaning [23].
Contaminant Lactobacillus are a drain for available sugars that would be converted into ethanol by yeasts, as well as for nutrients needed for optimal yeast growth and ethanol production [57, 61]. Makanjuola et al. [57] deliberately added pure cultures of Lactobacillus brevis, L. plantarun or Leuconostoc spp. to a laboratory scale malt whisky fermentation, and observed reduced yields of ethanol ranging from 6 to 22 %, lower yeast crops, reduced carbohydrate utilization, increased acidity due to acid lactic production and lower final pH, as well as foam production and flocculation. Chang et al. [21] isolated bacteria from ethanol production from starch, identified them and studied the effect of these contaminants in laboratory scale-fermentations. Authors found that all isolated bacteria were lactic acid producing, and Lactobacillus fermentum, L. casei and L. salivarium were the predominant species. These bacteria led to a greater than 30 % reduction in ethanol production in a cell-recycled continuous process. Narendranath et al. [61], studied the effect of Lactobacillus plantarum, L. paracasei, L. rhamnosus, and L. fermentum in ethanol productivity, and found a 3.8–7.6 % reduction in ethanol concentration, depending on the contaminant strain. Lactobacillus also cause problems in the food industry, like deterioration or contamination of meat products, pickles, mayonnaise, salad dressing, cheese, soy sauce, sake and beer [48].
Comparison of bacterial richness and diversity and the number of OTUs present in the different ethanol production stages showed that the most diverse bacterial sample is mixed juice (Table 2). Good´s coverage values for sugarcane juice and clarified juice samples indicated that a higher number of sequences would be needed to cover their total diversity. The PCA graphs (Fig. 3) and AMOVA results demonstrated that bacterial communities differed significantly between samples, except for evaporated juice and must samples. This result was expected, because the main significant difference between samples of evaporated juice and must is temperature (i.e., 92 °C for evaporated juice, 35 °C for must).
Conclusions
In summary, our analysis of microbial biodiversity in the stages of sugarcane ethanol production revealed changes in the microbial community as the process advances. In the earliest step of ethanol production, sugarcane juice has high bacterial diversity, which further increases in the next stage (mixed juice). It is likely that these microorganisms enter the ethanol production process with feedstock and soil impurities. High-throughput sequencing revealed a lower diversity of archaea and fungi in these two early stages. The presence of archaea in the ethanol production process has not been previously reported. In the next steps of the process (i.e., clarified juice and evaporated juice), bacterial diversity decreases as temperature increases. The predominant bacteria in these stages are capable of producing biofilms and spores, which may explain their presence in these samples and the occurrence of re-infections in subsequent ethanol-producing seasons. Biofilm-producing bacteria were also predominant in must, likely because of the higher sugar concentration and lower temperature. However, after fermentation the wine is dominated by Lactobacillus and unclassified Lactobacillaceae, with almost 100 % of the sequences belonging to these groups. Lactobacillus was the most common genus throughout the ethanol production process. Although these bacteria are present from the beginning of the process, they appear to have selective advantage over other bacteria at the end of the process through their tolerance for ethanol [54, 71] and production of acids that can kill other bacteria and yeasts. Thus, it seems that the process itself strongly selects for Lactobacillus. It will be interesting to further study these Lactobacillus populations to determine which specific ones are present.
The present study is the first to reveal the presence of archaea in sugarcane juice and mixed juice used for industrial ethanol fermentation. Our analysis showed that despite many studies investigating contaminants of ethanol production, there is still a lot to be learned about the diversity of microorganisms associated with this process, with many of the microbes designated as unclassified Archaea, Bacteria, and Fungi. The microbial diversity found in this work was higher than that described by previous studies. However, additional research is needed to determine how contaminant microbial communities may change over time in the same industrial facility, and whether they differ among sugarcane ethanol plants in the same geographical region. Furthermore, new studies should address how antibiotics commonly used in industry to control contaminants affect these microbial communities.
References
Abdullah SK, Saleh YA (2010) Mycobiota associated with sugarcane (Saccharum officinarum L.) cultivars in Iraq. Jordan J Biol Sci 3(4):193–202
Abdullah SK, Saleh YA (2010) Mycobiota associated with sugarcane (Saccharum officinarum L.) in Iraq. Mitosporic fungi. J Duhok Univ 13(1):130–138
Alcarde AR, Walder JMM, Horii J (2003) Fermentation of irradiated sugarcane must. Sci Agric (Piracicaba, Braz) 60(4):677–681
Basílio ACM, de Araújo PRL, de Morais JOF, da Silva Filho EA, de Morais MA, Simões DA (2008) Detection and identification of wild yeast contaminants of the industrial fuel ethanol fermentation process. Curr Microbiol 56(4):322–326. doi:10.1007/s00284-007-9085-5
Bayrock DP, Ingledew WM (2004) Inhibition of yeast by lactic acid bacteria in continuous culture: nutrient depletion and/or acid toxicity? J Ind Microbiol Biotechnol 31(8):362–368. doi:10.1007/s10295-004-0156-3
Bayrock DP, Thomas KC, Ingledew WM (2003) Control of Lactobacillus contaminants in continuous fuel ethanol fermentations by constant or pulsed addition of penicillin G. Appl Microbiol Biotechnol 62:498–502. doi:10.1007/s00253-003-1324-5
Beckner M, Ivey ML, Phister TG (2011) Microbial contamination of fuel ethanol fermentations. Lett Appl Microbiol 53:387–394. doi:10.1111/j.1472-765X.2011.03124.x
Bhothipaksa K, Busta FF (1978) Osmotically induced increase in thermal resistance of heat-sensitive, dipicolinic acid-less spores of Bacillus cereus Ht-8. Appl Environ Microbiol 35(4):800–808
Bintrim SB, Donohue TJ, Handelsman J, Roberts GP, Goodman RM (1997) Molecular phylogeny of Archaea from soil. Proc Natl Acad Sci USA 94(1):277–282
Bischoff KM, Liu S, Leathers TD, Worthington RE, Rich JO (2009) Modeling bacterial contamination of fuel ethanol fermentation. Biotechnol Bioeng 103(1):117–122. doi:10.1002/bit.22244
BNDES, CGEE (2008) Bioetanol de cana-de-açúcar: energia para o desenvolvimento sustentável, 1st ed. BNDES, Rio de Janeiro
Boddey R, Urquiaga S, Alves BR, Reis V (2003) Endophytic nitrogen fixation in sugarcane: present knowledge and future applications. Plant Soil 252(1):139–149. doi:10.1023/a:1024152126541
Brady CL, Venter SN, Cleenwerck I, Vandemeulebroecke K, De Vos P, Coutinho TA (2009) Transfer of Pantoea citrea, Pantoea punctata and Pantoea terrea to the genus Tatumella emend. as Tatumella citrea comb. nov., Tatumella punctata comb. nov. and Tatumella terrea comb. nov. and description of Tatumella morbirosei sp. nov. Int J Syst Evol Microbiol 60(3):484–494. doi:10.1099/ijs.0.012070-0
Brasil (2014) Boletim mensal dos combustíveis renováveis. Janeiro/2014. Ministério de Minas e Energia (MME), Brasília
Brasil (2014) Relação das unidades produtoras cadastradas no Departamento da Cana-de-açúcar e Agroenergia (Maio/14). Ministério da Agricultura, Pecuária e Abastecimento (MAPA), Brasília
Brochier-Armanet C, Gribaldo S, Forterre P (2011) Spotlight on the Thaumarchaeota. ISME J 6(2):227–230. doi:10.1038/ismej.2011.145
Cabrini KT, Gallo CR (1999) Identificação de leveduras no processo de fermentação alcoólica em usina no estado de São Paulo, Brasil. Sci Agric (Piracicaba, Braz) 56(1):207–216
Camolez MA, Mutton MJR (2005) Influência de microrganismos contaminantes sobre o processo fermentativo. STAB 23(5):6–9
Cao P, Zhang L-M, Shen J-P, Zheng Y-M, Di HJ, He J-Z (2012) Distribution and diversity of archaeal communities in selected Chinese soils. FEMS Microbiol Ecol 80(1):146–158. doi:10.1111/j.1574-6941.2011.01280.x
Castro MMdS (1995) Leveduras contaminantes do processo de fermentação alcoólica: diversidade taxonômica e metabólica. Dissertação, Universidade estadual de campinas, Campinas
Chang I-S, Byung-Hong K, Pyong-Kyun S, Wan-Kyu L (1995) Bacterial contamination and its effects on ethanol fermentation. J Microbiol Biotechnol 5(6):304–314
Chang IS, Kim BH, Shin PK (1997) Use of sulfite and hydrogen peroxide to control bacterial contamination in ethanol fermentation. Appl Environ Microbiol 63(1):1–6
Cherubin RA (2003) Efeitos da viabilidade da levedura e da conaminação bacteriana na fermentação alcoólica. Tese, Escola Superior de Agricultura Luiz de Queiroz, Piracicaba
Chieppe Júnior JB (2012) Tecnologia e fabricação do álcool. Universidade Federal de Santa Maria, Inhumas
Conab (2012) Perfil do setor do açúcar e do álcool no Brasil—Edição para a safra 2009–2010. Companhia Nacional de Abastecimento (Conab), Brasília
Conab (2014) Acompanhamento de safra brasileira: cana-de-açúcar, quarto levantamento, abril/2014. Companhia Nacional de Abastecimento (Conab), Brasília
Cunha IS, Barreto CC, Costa OYA, Bomfim MA, Castro AP, Kruger RH, Quirino BF (2011) Bacteria and Archaea community structure in the rumen microbiome of goats (Capra hircus) from the semiarid region of Brazil. Anaerobe 17(3):118–124. doi:10.1016/j.anaerobe.2011.04.018
David V, Terrat S, Herzine K, Claisse O, Rousseaux S, Tourdot-Maréchal R, Masneuf-Pomarede I, Ranjard L, Alexandre H (2014) High-throughput sequencing of amplicons for monitoring yeast biodiversity in must and during alcoholic fermentation. J Ind Microbiol Biotechnol 41(5):811–821. doi:10.1007/s10295-014-1427-2
de Azeredo LA, A. GE, Mendonca-Hagler LC, Hagler AN (1998) Yeast communities associated with sugarcane in Campos, Rio de Janeiro, Brazil. Int Microbiol 1(3):205–208
de Carvalho GG, Monteiro RAB (2011) A influência do teor de acidez e da contaminação bacteriana do mosto no rendimento fermentativo industrial para produção de etanol. FAZU 8:47–54
de Souza Liberal AT, da Silva Filho EA, de Morais JO, Simoes DA, de Morais MA, Jr (2005) Contaminant yeast detection in industrial ethanol fermentation must by rDNA-PCR. Lett Appl Microbiol 40(1):19–23
DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci USA 89(12):5685–5689
Dicks LMT, Van Vuuren HJJ, Dellaglio F (1990) Taxonomy of Leuconostoc species, particularly Leuconostoc oenos, as revealed by numerical analysis of total soluble cell protein patterns, DNA base compositions, and DNA–DNA hybridizations. Int J Syst Bacteriol 40(1):83–91. doi:10.1099/00207713-40-1-83
Dong Z, Heydrich M, Bernard K, McCully ME (1995) Further evidence that the N(inf2)-fixing endophytic bacterium from the intercellular spaces of sugarcane stems is Acetobacter diazotrophicus. Appl Environ Microbiol 61(5):1843–1846
Gallo CR (1989) Determinação da microbiota bacteriana de mosto e de dornas de fermentação alcoólica. Universidade Estadual de Campinas, São Paulo, Tese
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes–application to the identification of mycorrhizae and rusts. Mol Ecol 2(2):113–118
Goldemberg J, Macedo IC (1994) Brazilian alcohol program. An overview. Energy Sust Dev 1(1)
Granhall U, Welsh A, Throbäck IN, Hjort K, Hansson M, Hallin S (2010) Bacterial community diversity in paper mills processing recycled paper. J Ind Microbiol Biotechnol 37(10):1061–1069. doi:10.1007/s10295-010-0754-1
Grosskopf R, Janssen PH, Liesack W (1998) Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl Environ Microbiol 64(3):960–969
Hansel CM, Fendorf S, Jardine PM, Francis CA (2008) Changes in Bacterial and Archaeal community structure and functional diversity along a geochemically variable soil profile. Appl Environ Microbiol 74(5):1620–1633. doi:10.1128/aem.01787-07
Hoon C, Park S-Y, Ryu C-M, Kim JF, Park S-H, Park CS (2005) Diversity of root-associated Paenibacillus spp. in winter crops from the southern part of Korea. J Microbiol Biotechnol 15(6):1286–1298
Hu H-W, Zhang L-M, Yuan C-L, He J-Z (2013) Contrasting Euryarchaeota communities between upland and paddy soils exhibited similar pH-impacted biogeographic patterns. Soil Biol Biochem 64:18–27. doi:10.1016/j.soilbio.2013.04.003
Hynes SH, Kjarsgaard DM, Thomas KC, Ingledew WM (1997) Use of virginiamycin to control the growth of lactic acid bacteria during alcohol fermentation. J Ind Microbiol Biotechnol 18(4):284–291
Ignatova-Ivanova T, Ivanov R (2013) Anticorrosion effect of biofilm forming by Lactobacillus strains on metal surfaces. Bulg J Agric Sci 19(2):83–85
Imhoff JF (2005) Enterobacteriales. In: Brenner D, Krieg N, Staley J et al (eds) Bergey’s manual® of systematic bacteriology. Springer US, pp 587–850. doi:10.1007/0-387-28022-7_13
Karner MB, DeLong EF, Karl DM (2001) Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature (London) 409(6819):507–510. doi:10.1038/35054051
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk PM, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüßler A, Scott JA, Senés C, Smith ME, Suija A, Taylor DL, Telleria MT, Weiss M, Larsson K-H (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22(21):5271–5277. doi:10.1111/mec.12481
Kubota H, Senda S, Nomura N, Tokuda H, Uchiyama H (2008) Biofilm formation by Lactic Acid Bacteria and resistance to environmental stress. J Biosci Bioeng 106(4):381–386. doi:10.1263/jbb.106.381
Lane DJ (1991) 16S/23S sequencing. In: Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York
Leja K, Broda M (2009) The occurrence and identification of microbiological contamination in fuel ethanol production. Acta Sci Pol 8(4):25–31
Limtong S, Kaewwichian R, Yongmanitchai W, Kawasaki H (2014) Diversity of culturable yeasts in phylloplane of sugarcane in Thailand and their capability to produce indole-3-acetic acid. World J Microbiol Biot 30(6):1785–1796. doi:10.1007/s11274-014-1602-7
Loiret FG, Ortega, Kleiner D, Ortega-Rodes P, Rodes R, Dong Z (2004) A putative new endophytic nitrogen-fixing bacterium Pantoea sp. from sugarcane. J Appl Microbiol 97(3):504–511
Lozupone C, Hamady M, Knight R (2006) UniFrac-an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform 7:371. doi:10.1186/1471-2105-7-371
Lucena BT, dos Santos BM, Moreira JL, Moreira AP, Nunes AC, Azevedo V, Miyoshi A, Thompson FL, De Morais MA Jr (2010) Diversity of lactic acid bacteria of the bioethanol process. BMC Microbiol 10:298. doi:10.1186/1471-2180-10-298
Madigan MT, Martinko JM, Parker J (2004) Brock, Biología de Los Microorganismos, 10th edn. Pearson Prentice Hall, Upper Saddle River
Makanjuola DB, Springham DG (1984) Identification of lactic acid bacteria isolated from different stages of malt whisky distillery fermentations. J Inst Brew 90:13–19
Makanjuola DB, Tymon A, Springham DG (1992) Some effects of lactic acid bacteria on laboratory-scale yeast fermentations. Enzyme Microb Tech 14(5):350–357. doi:10.1016/0141-0229(92)90002-6
Marteinsson VT, Birrien JL, Reysenbach AL, Vernet M, Marie D, Gambacorta A, Messner P, Sleytr UB, Prieur D (1999) Thermococcus barophilus sp. nov., a new barophilic and hyperthermophilic archaeon isolated under high hydrostatic pressure from a deep-sea hydrothermal vent. Int J Syst Bacteriol 49(2):351–359
Merlino G, Rizzi A, Villa F, Sorlini C, Brambilla M, Navarotto P, Bertazzoni B, Zagni M, Araldi F, Daffonchio D (2012) Shifts of microbial community structure during anaerobic digestion of agro-industrial energetic crops and food industry byproducts. J Chem Technol Biotechnol 87(9):1302–1311. doi:10.1002/jctb.3784
Murray RG, Stackebrandt E (1995) Taxonomic note: implementation of the provisional status Candidatus for incompletely described procaryotes. Int J Syst Bacteriol 45(1):186–187
Narendranath NV, Hynes SH, Thomas KC, Ingledew WM (1997) Effects of Lactobacilli on yeast-catalyzed ethanol fermentations. Appl Environ Microbiol 63(11):4158–4163
TdP Nobre, Horii J, Alcarde AR (2007) Viabilidade celular de Saccharomyces cerevisiae cultivada em associação com bactérias contaminantes da fermentação alcoólica. Ciên Tecnol Alim 27:20–25
Perin L, Baldani JI, Reis VM (2004) Diversidade de Gluconacetobacter diazotrophicus isolada de plantas de cana-de-açúcar cultivadas no Brasil. Pesq Agropec Bras 39:763–770
Pester M, Schleper C, Wagner M (2011) The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology. Curr Opin Microbiol 14(3):300–306. doi:10.1016/j.mib.2011.04.007
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:590–596. doi:10.1093/nar/gks1219
Quecine MC, l. AW, Rossetto PB, Ferreira A, Tsui S, Lacava PT, Mondin M, Azevedo JL, Pizzirani-Kleiner AA (2012) Sugarcane growth promotion by the endophytic bacterium Pantoea agglomerans. Appl Environ Microbiol 78(21)
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ (2011) Removing noise from pyrosequenced amplicons. BMC Bioinform 12(1):38. doi:10.1186/1471-2105-12-38
Rennie RJ, Freitas JRd, Ruschel AP, Vose PB (1982) Isolation and identification of N2-fixing bacteria associated with sugar cane (Saccharum sp.). Can J Microbiol 28(5):462–467. doi:10.1139/m82-070
RFA (2013) World Fuel Ethanol Production. Renewable Fuels Association. http://ethanolrfa.org/pages/World-Fuel-Ethanol-Production. Accessed 25.07.2014
Rich JO, Leathers TD, Nunnally MS, Bischoff KM (2011) Rapid evaluation of the antibiotic susceptibility of fuel ethanol contaminant biofilms. Bioresour Technol 102(2):1124–1130. doi:10.1016/j.biortech.2010.08.118
Salminen S, Av Wright, Lahtinen S, Ouwehand A (2012) Lactic acid bacteria: microbiological and functional aspects, 4th edn. Marcel Dekker, New York
Santoro AE, Casciotti KL (2011) Enrichment and characterization of ammonia-oxidizing archaea from the open ocean: phylogeny, physiology and stable isotope fractionation. ISME J 5(11):1796–1808
Schleper C, Puhler G, Kuhlmorgen B, Zillig W (1995) Life at extremely low pH. Nature (London) 375(6534):741–742. doi:10.1038/375741b0
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Ka Skinner-Nemec, Nichols NN, Leathers TD (2007) Biofilm formation by bacterial contaminants of fuel ethanol production. Biotechnol Lett 29:379–383. doi:10.1007/s10529-006-9250-0
Skinner KA, Leathers TD (2004) Bacterial contaminants of fuel ethanol production. J Ind Microbiol Biotechnol 31(9):401–408. doi:10.1007/s10295-004-0159-0
Stahl DA, Amann RI (1991) Development and application of nucleic acid probes in bacterial systematics In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York
Thomas KC, Hynes SH, Ingledew WM (2001) Effect of lactobacilli on yeast growth, viability and batch and semi-continuous alcoholic fermentation of corn mash. J Appl Microbiol 90(5):819–828
Timmusk S, Grantcharova N, Wagner EGH (2005) Paenibacillus polymyxa invades plant roots and forms biofilms. Appl Environ Microbiol 71(11):7292–7300. doi:10.1128/aem.71.11.7292-7300.2005
Tortora GJ, Funke BR, Case CL (2012) Microbiologia, 10th edn. Artmed, Porto Alegre
Turner S, Pryer KM, Miao VPW, Palmer JD (1999) Investigating deep phylogenetic relationships among Cyanobacteria and plastids by small subunit rRNA sequence analysis. J Euk Microbiol 46(4):327–338. doi:10.1111/j.1550-7408.1999.tb04612.x
UNICA (2014) Maior produtor mundial de cana-de-açúcar. União da indústria de cana-de-açúcar. http://www.unica.com.br/faq/. Accessed 24 July 2014
White J, Gilbert J, Hill G, Hill E, Huse SM, Weightman AJ, Mahenthiralingam E (2011) Culture-independent analysis of bacterial fuel contamination provides insight into the level of concordance with the standard industry practice of aerobic cultivation. Appl Environ Microbiol 77(13):4527–4538. doi:10.1128/aem.02317-10
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego
Whitehead TR, Cotta MA (1999) Phylogenetic diversity of methanogenic archaea in swine waste storage pits. FEMS Microbiol Lett 179(2):223–226. doi:10.1016/S0378-1097(99)00415-2
Wright A-DG, Ma X, Obispo NE (2007) Methanobrevibacter phylotypes are the dominant methanogens in sheep from venezuela. Microb Ecol 56(2):390–394. doi:10.1007/s00248-007-9351-x
Zhu XY, Lubeck J, Kilbane JJ (2003) Characterization of microbial communities in gas industry pipelines. Appl Environ Microbiol 69(9):5354–5363. doi:10.1128/aem.69.9.5354-5363.2003
Acknowledgments
O.Y.A.C. was supported by a CNPq fellowship. This work was supported by grant from CNPq, FAD-DF, Embrapa.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Costa, O.Y.A., Souto, B.M., Tupinambá, D.D. et al. Microbial diversity in sugarcane ethanol production in a Brazilian distillery using a culture-independent method. J Ind Microbiol Biotechnol 42, 73–84 (2015). https://doi.org/10.1007/s10295-014-1533-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1533-1