
Abstract
IgG4-related disease (IgG4-RD) is still an underestimated disorder which affects multiple organs, and its recognition as a distinct clinical disease has been only proved in the recent decades. The renal involvement has been documented in approximately 15% of patients with IgG4-RD, and the typical manifestation is a tubulo-interstitial nephritis. The main histological findings in IgG4-RD are typically a dense tissue infiltration of IgG4-positive plasma cells, storiform fibrosis, obliterative phlebitis, and frequently elevated IgG4 serum levels. Herein we report our atypical and peculiar clinical presentation of an IgG4-related nephropathy (IgG4-RN) and the remarkable response to rituximab (RTX) treatment at the renal imaging with computerized tomography assessment. The current nephrological evidences support the renal function recovery after steroids or steroids plus RTX therapy, even if the renal imaging data are not always shown. In a complex and enigmatic clinical scenario such as the IgG4-RN, both the renal biopsy and the renal imaging before and after the immunosuppressive therapy become mandatory tools to thoroughly define the diagnosis, the management and the response to the immunological therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IgG4-related disease (IgG4-RD) is underestimated and challenging to diagnose disorder, which affects multiple organs and thus has been recognized as a distinct clinical entity in the recent decades [1]. IgG4-RD is typically characterized by dense tissue infiltration of IgG4-positive plasma cells, storiform fibrosis, obliterative phlebitis, and frequently elevated IgG4 serum levels [2, 3].
IgG4-RD was firstly identified in 1961 as a sclerosing pancreatic lesion occurring in autoimmune pancreatitis [4, 5]. However, similar extrapancreatic inflammatory lesions have been described also in patients without pancreatic disease [6, 7].
Although IgG4-RD usually affects more than one organ either simultaneously or sequentially, single organ involvement, such as kidney-only disease, has occurred [8, 9]. Any renal disease in IgG4-RD is collectively considered as IgG4-related nephropathy (IgG4-RN), and it has been observed in approximately 15% of patients with IgG4-RD [10, 11].
In this paper, we describe a remarkable case of IgG4-RN with impressive macroscopic renal presentation but with preserved renal function.
Case presentation
A 57-year-old Caucasian male was admitted to our Renal Unit in February 2019 due to persistent fever with chills for almost 1 month. Both blood and urine cultures were negative, and his renal function was normal (serum creatinine 0.96 mg/dL, estimated glomerular filtration rate 87.4 ml/min/1.73 m2 according to the CKD-EPI formula. See Table 1 for complete blood and urine examinations).
His medical history reported that he had been suffering from ulcerative recto-colitis since 2010. In August of 2012, he was admitted to another hospital because of ground glass pneumonia with pleural effusion and over- and sub-diaphragmatic lymphadenopathy. At that time, both pleural drainage and supraclavicular lymphnode biopsy were performed; the former exam was positive for Staphylococcus aureus infection, while the latter was negative for lymphoproliferative disorders and/or granulomatosis. Renal function was normal, and the patient was treated with an antibiotic regimen based on tigecycline due to the antibiogram result. Once the therapy was concluded, he was discharged with a presumed diagnosis of Castleman's disease.
In October of 2012, he was admitted to another hospital due to the onset of an acute renal injury (serum creatinine 1.9 mg/dL) with gross hematuria and nephrotic proteinuria (4.2 gr/die). A renal biopsy showed an exudative and membrano-proliferative glomerulonephritis associated with tubule-interstitial nephritis and undefined granulomatosis, potentially attributable to Castleman’s disease or Sarcoidosis. Prednisone of unreported dose was prescribed, and the patient recovered from the acute renal injury, enabling him to be discharged from the hospital with a serum creatinine of 0.7 mg/dL. He was prescribed a therapy of: 15 mg of methotrexate once a week, 25 mg/day of prednisone, 5 mg/day of ramipril and 20 mg/day of omeprazole, and he was referred to a rheumatology outpatient clinic for a presumed “Castleman-like/sarcoidosis” syndrome with stable and normal renal function across the years.
His home therapy since 2013 included 5 mg/day of prednisone, 2.5 mg/week of methotrexate and a single 20 mg tablet/day of esomeprazole.
Upon admission to our hospital in February 2019, the patient underwent both lung and abdominal contrast computer-assisted tomography (CT), which showed a markedly enlarged left kidney (longitudinal diameter of 14 cm) with diffuse hypodense areas, absence of cortico-medullary differentiation and a poorly visualized pyelocalyceal system (see Fig. 1). The right kidney was also affected, but with a less severe presentation of multiple cortical hypodense formations with indistinct margins. Renal function was normal during the entire time that he was hospitalized and the patient recovered from fever after antibiotic therapy with 2 g/day of ceftriaxone for three weeks.

Abdominal CT scan. CT images obtained through the abdomen and pelvis with intravenous iodinated contrast demonstrate a severely increased left kidney (craniocaudal length 14 cm) with associated complete parenchymal structural alteration due to the presence of diffuse hypodense areas without corticomedullary differentiation and poorly visualized pyelocalyceal system. On the left, the renal artery and posterior wall of the renal vein are surrounded by a 20-mm-thick sleeve of hypodense tissue. There is associated mild thickening of the perirenal fascia and no evidence of contrast medium excretion even during the delayed urographic phase. The described findings are not univocal and clinical-laboratory correlation and histological examination are needed (renal granulomatous involvement in sarcoidosis? Primary tumor? Inflammation due to other causes?). The right kidney is normal in size (craniocaudal length 11 cm), shape and nephrogram; it is characterized by the presence of multiple cortical hypodense formations with indistinct margins (maximum diameter of 20 mm at the level of anterior peri-hilar region). This could be interpreted as an initial manifestation of the same disease process. There is no evidence of focal pathological alterations at the level of the remaining abdominal parenchymal organs. Pelvis cannot be fully evaluated due to artifacts from right hip prosthesis; there is no evidence of abdominal ascites or significant lymphadenopathy, except for some subcentrimetric lymph nodes in the left para-aortic region
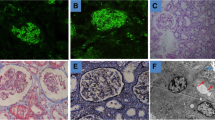
Due to the uncertainty of his renal condition, a second renal biopsy of the left kidney was performed with the support of an interventional radiologist. The procedure was manageable preventing bleeding and other complications (see Fig. 2). The biopsy specimen resulted mostly of connective tissue without a clear distinction between renal cortex and medulla. Three out of eight glomeruli showed a hyaline morphology with various degrees of tubular damage. In the other glomeruli, thickening of the Bowman capsule and of the basement membrane was observed with normal mesangial matrix. In two glomeruli, segmental collapse of the capillary vessels was also detected. A severe tubular-interstitial nephritis with numerous IgG4 + plasma cells and extensive interstitial storiform fibrosis was identified at light microscopy (see Fig. 2).

Renal histological findings at light microscopy. The biopsy specimen resulted mostly of connective tissue without a clear distinction between renal cortex and medulla. In this setting, 3 out of 8 glomeruli showed a hyaline morphology with various degree of tubular deterioration. In the other glomeruli, thickening of the Bowman capsule and of the basement membrane was observed with normal mesangial matrix. In two glomeruli, the segmental collapse of the capillary vessels was also highlighted. a Severe tubulointerstitial nephritis with numerous IgG4 plasma cells and expansile interstitial fibrosis was identified at the renal biopsy. b 15–60 IgG4 + plasma cells per 40X magnification field were present with interspersed mast cells. Severe tubular atrophy can also be detected
IgG subclasses were dosed in the serum with non-significant results: IgG1 415 mg/dL, normal values (n.v.) 490–1.140 mg/dL; IgG2 194 mg/dL, n.v. 105–640 mg/dL; IgG3 17 mg/dL, n.v. 20–110 mg/dL; IgG4 96 mg/dL n.v. 8–140 mg/dL.
A therapy of rituximab 375 mg/m2/week for 4 weeks and associated prednisone 0.5 mg/kg/day was prescribed in accordance with a diagnosis of IgG4-RN.
6 months later the patient underwent another abdominal contrast-enhanced CT scan that showed a remarkable reduction in longitudinal diameter of the left kidney (4 cm vs 14 cm) with consistent and diffuse hypodensity (see Fig. 3). The right kidney had significantly improved since hypodense areas were not any more appreciated. Currently, the patient proceeds his follow-up in the outpatients’ glomerulonephritis clinic and no other admissions to hospital have been occurred.

Abdominal contrast-enhanced CT scan after 6 Months. Hypotrophic left kidney markedly reduced in diameter compared to previous CT (dimensions 4 × 4 vs. 14 × 8, 5 cm) with uniform contrast enhancement reduction in each phase. Persistent dense area of 20 mm in the renal hilus. Normal morphology and diameter of the right kidney with preserved contrast enhancement. A simple cortical cysts with maximum diameter of 11.6 mm is present at the §§§. The hypodense areas of the renal parenchyma previously described and compatible with IgG4-related nephropathy were no more present
Discussion
IgG4-RN is the renal involvement of IgG4-RD that could show a wide spectrum of clinical and histological features including tubulointerstitial nephritis (TIN), membranous glomerulonephritis (MGN), pyelitis and hydronephrosis as a consequence of retroperitoneal fibrosis (RPF) [9].
The most common presentation of the disease is IgG4-related TIN, and its histological hallmarks are: a dense lymphoplasmacytic infiltrate enriched with IgG4 + plasma cells, and a variable degree of storiform fibrosis [7], (see Table 2). Often IgG4-TIN shows the presence of Mott cells (plasma cells that have spherical immunoglobulin inclusions packed in their cytoplasm) in the interstitium. Interstitial eosinophylia can also be observed and may lead to misclassification of allergic TIN [12]. Tubular basement membrane may also exhibit immune complex deposits of IgG, especially at the trichrome stain. Also kappa and lambda light chains and even C3 deposits have been described [7,8,9].
Obliterative phlebitis, which is another hallmark of IgG4-RD due to the presence of a dense lymphoplasmacytic infiltrate in the venous vessels, is not always appreciated in organs such as the salivary gland, lymph node and the kidney [13, 14]. This could be explained by the limitations of the needle biopsy involving such organs to show veins [13, 14].Hara et al. [15] reported that IgG4-TIN also tends to involve the perivascular areas of medium and small-sized vessels in the renal cortex with inflammation or fibrosis.
According to the Mayo Clinic classification of IgG4-TIN, three patterns can be described: pattern A is characterized by acute TIN with minimal interstitial fibrosis; pattern B shows chronic TIN with expansive interstitial fibrosis; while pattern C involves advanced sclerosing form with fewer inflammatory cells [16]. The typical clinical presentations of patients with IgG4-TIN are kidney mass lesions and/or renal insufficiency [17]. In our patient, the histological examination revealed pattern B of IgG4-TIN and a renal mass presentation but without renal failure.
MGN is the most common glomerular involvement in IgG4-RN, and it occurs in approximately 7% of IgG4-RN [9]. As in primary MGN, the IgG4 subclass is the dominant immunoglobulin in the glomerular immune deposits in IgG4-related MGN, while serum anti-M-type phospholipase A2 receptor antibody is negative. The clinical presentation is generally characterized by a nephrotic-range proteinuria[9].
RPF is a chronic inflammatory and fibrotic disease involving periaortic and peri-iliac retroperitoneal tissue, often leading to hydroureteronephrosis when urine flow is obstructed at the ureters [18]. IgG4-RD is now identified as the cause of up to two-thirds of the cases of idiopathic RPF [19].
Because of the variable expression of this disease, a differential diagnosis that takes into account other immunological disorders is required. These other disorders are: allergic TIN, Sjögren syndrome, granulomatosis with polyangiitis, Castleman disease, and sarcoidosis that may involve the kidneys (see Table 3).
Patients affected by IgG4-RD and/or IgG4-RN show high levels of IgG in 70–90% of the cases. An elevation of IgG4 is present in 50–70% of the patients, although IgG4 is not specific to IgG4-RD and a high level could also be observed in other diseases such as biliary and pancreatic disorders.20 Other prominent laboratory findings are elevated serum IgE levels (60–70%), C3 and/or C4 hypocomplementemia (50–70%), peripheral eosinophilia (35–50%), antinuclear antibody (30%) and rheumatoid factor (30%) [20, 21].
In patients with only a slight increase in IgG4 levels, specificity may be enhanced by higher ratios of IgG4/IgG > 10% or IgG4/IgG1 > 24%, but there is no consensus regarding this issue in the literature [20].
The radiological assessment of IgG4-RN is often incidental, and both abdominal ultrasound and contrast-enhanced computed tomography (CT) are the preferred imaging tools to evaluate patients with suspected IgG4-RN. On ultrasound, unilateral or bilateral kidney swelling could be observed even with hydroureteronephrosis when RPF is present [7].
As can be seen with a CT scan, IgG4-RN could present itself in various manners, which include: multiple unilateral or bilateral small hypodense lesions, diffuse heterogeneous enhancement of the kidney, soft tissue changes surrounding the kidneys or by well-defined hypodense exophytic mass(es) [21]. Renal involvement may rarely manifest as a solitary lesion [19]. These findings may mimic both immunological and malignant diseases and renal biopsy should be considered in the diagnostic process to rule out the possibility of IgG4-RN.
Though magnetic resonance (MRI), renal lesions may appear hypointense on both T1- and T2-weighted images compared to the normal renal cortex and may show mild contrast enhancement after gadolinium administration [22]. Recently carried out diffusion-weighted MRI also showed 100% sensitivity for IgG4-RN [22].
Our clinical case reported multiple hypodense lesions in both kidneys on computer tomography according to the most common macroscopic presentation of IgG4-related nephropathy (IgG4-RN) that shows a multiple low-density lesions scenario [9], that has been appreciated in almost 65% of IgG4-related kidney disease patients [21]. Our patient exhibited a markedly enlarged left kidney (longitudinal diameter of 14 cm) with diffuse hypodense areas and also the right kidney showed initial involvement with different hypodense areas, even if with a less severe presentation compared to the contralateral. In addition, we should consider that our patient was already on immunosuppressive therapy since 2013, in fact he was on a low dose of prednisone (5 mg/day) and 2.5 mg/day of methotrexate after being treated with 15 mg of methotrexate once a week, 25 mg/day of prednisone, 5 mg/day of ramipril following the first renal biopsy on 2012.
In this setting, how the impact of the previous immunological treatments may have conditioned the renal involvement and the onset of the macroscopic lesions is hard to assess. However, according to the fact that our patient was already on a steroid treatment we decided to consider an immunosuppressive therapy based non only on a steroid regimen.
The management of IgG4-RN is similar to that of IgG4-RD, with emphasis on the use of glucocorticoids as the first line of therapy [23, 24], as recommended by the 2015 International Consensus Statement Guidance on the management and Treatment of IgG4-RD [25]. The typically adopted therapy regimen comprises prednisone 0.6–1 mg/kg/day, which is gradually tapered after 2–4 weeks to induce remission. This therapeutic strategy has been associated with a response rate higher than 80% [26]. Various studies have attempted to optimize the reduction of prednisone to have the highest efficiency possible [27, 28].One scenario is to initially decrease the daily dose by 10 mg every 2 weeks until a daily dose of 20 mg is reached, and keep that same dosage for a short time of treatment (e.g., 2 weeks) and then subsequently resume the dose reduction, this time by 5 mg every 2 weeks. According to many authors, the goal of the induction therapy is to withdraw glucocorticoid 3–6 months after the start of treatment [29, 30]. On the contrary, many Japanese authors recommend the use of low-dose glucocorticoid maintenance therapy for up to 3 years [31].
Saeki et al. [9] reported in a study of 19 Japanese patients affected by IgG4-RN, a rapid improvement in 18 patients (94.7%) of both renal function and renal radiological findings even at 1 month after the start of steroid therapy (oral prednisolone 0.6 mg/kg/day as induction therapy for 2–4 weeks and then gradually tapered to a low-dose maintenance dose). Misushima et al. [32] also reported quickly amelioration of renal lesions on computer tomography after corticosteroid therapy in six patients affected by IgG4-RN. In fact, three patients showed recovery of contrast enhancement of the renal cortex and two patients exhibited improvement of renal swelling after steroid therapy, although scar-like focal cortical atrophy persisted in two patients. These atrophic lesions in imaging studies may be the macroscopic presentation of renal fibrosis areas, less responsive to steroid treatment. Also Mise et al. [33] reported an impressive and effective impact of prednisone therapy (from 60 mg/day to 2 mg/day over 4 years) on both renal function and morphology on markedly enlarged kidneys with irregular contrast staining due to IgG4-RN. However, the last authors did not report the kidney diameters before and after the steroid therapy, but they did not report any disease recurrence across the following four years.
In addition, a prolonged steroid-based treatment should always be carefully evaluated and monitored because it could be associated with adverse side effects like new onset of or worsening of diabetes, acute cardiovascular events and osteoporosis.
A monoclonal antibody-based regimen with rituximab (RTX) has shown good results in improving clinical and serological features of IgG4-RD patients even without concomitant steroid therapy [34], leading to a decrease in serum IgG4 concentrations and better disease control. Unfortunately, the relapse of renal dysfunction following steroid discontinuation is frequently observed in IgG4-RD with kidney involvement [10, 35]. In this setting, a RTX-based regimen might offer a longer-lasting response even in IgG4-RN patients.
Caruthers et al. [34] reported RTX as a successful therapeutic agent in a 12-month, single-arm pilot trial of 30 IgG4-RD patients, 20% of whom showed different types of renal conditions, mostly tubulointerstitial nephritis. All patients received two IV doses of RTX 1000 mg, administered approximately 15 days apart, after the infusion of methylprednisolone 100 mg. Nearly 90% of the patients in this trial were treated with RTX alone, and disease response was observed in 97% of the patients at 6 months. 60% of patients (18 out of 30) experienced complete remission of the disease, at any time over the 12-month follow-up period, with 47% of cases within the first 6-month period.
During the course of the trial, only 4 patients experienced IgG4-RD flares with urgent manifestations: out of the two patients at three months, one exhibited pancreatic conditions and the other had carotid artery involvement. One patient at ten months had implications in the bile ducts, and one at twelve months exhibited affected lungs.
Several monocentric experiences regarding the association of steroids and RTX are reported in the literature [19, 35, 36]. Yamamoto et al. reported that using 500 mg/body of RTX plus initial dose of prednisone 30–40 mg/day on the onset of IgG4-RD relapse did not guarantee a complete remission and IgG4-RD patients experienced one or more relapses after RTX administration. Therefore, one or two administration of RTX could not induce or maintain complete remission of IgG4-RD for long.
Khosroshahi et al. [36] reported very different findings. In their ten patients with IgG4- RD, RTX was used (2 infusions of 1000 mg, 15 days apart) and six of the ten patients were also on chronic corticosteroid treatment. Following the administration of RTX, the baseline prednisone dose was slowly reduced in all patients in order to avoid adrenal insufficiency (the mean time to reach prednisone discontinuation was 5.3 months with a range of 1–9 months). Nine of the ten patients demonstrated substantial clinical improvement within 1 month of RTX therapy. Only two patients suffered disease recurrences approximately 6 months after RTX treatment and experienced a total of 4 disease flares between them. All disease flares promptly responded to repeat courses of RTX. Only one patient required glucocorticoid treatment.
Roccatello et al. treated 5 patients with IgG4-RN with the same regimen adopted for severe systemic lupus erythematosus (SLE) that consists of three pulses of 15 mg/kg intravenous methylprednisolone followed by oral tapering prednisone and two IV pulses of cyclophosphamide 500 mg (on days 1 and 15) and 4 weekly RTX administrations (375 mg/m2) [19, 37]. All patients experienced renal function improvement and those who repeated renal biopsy after 1 year showed a net reduction of tubulointerstitial infiltration as well as the disappearance of IgG4-positive plasma cells, but still with persistent fibrosis.
Hart and colleagues reported an 83% rate of complete remission following RTX treatment (3 weekly doses of 375 mg/m2) in a group of 12 IgG4-RD patients whose disease had been resistant to, or who had contraindications to steroids or conventional steroid-sparing agents [38].
RTX has been also useful as maintenance therapy, but the optimal frequency and duration of treatment have not been clarified yet. When used as maintenance therapy, RTX is often administered on evidence of disease flare rather than at a predetermined time interval (e.g., every 6–12 months). This practice requires further evidence as suggested by the partial or no efficacy of RTX when used in cases of severely sclerosed organs [39, 40], and might vary depending on the potential risk of organ damage associated with a disease relapse [25].
In our case, we decided a rituximab based approach (375 mg/m2/week for 4 week) and associated prednisone 0.5 mg/kg/day due to the previous and unsuccessful steroid regimen and according to the previous evidences in the literature. Our patient after six months exhibited a marked reduction in the left kidney longitudinal diameter (> 50%, 4 cm vs. 14 cm) even if with uniform contrast enhancement reduction in each phase and a persistent dense area. Of note, normal morphology and diameter of the right kidney with preserved contrast enhancement were also assessed and this report supports the positive impact of the rituximab treatment in steroid-resistant IgG4-RN, in all the patients with contraindications to steroid treatment and in those IgG4-RN patients with less severe renal fibrosis.
In conclusion, extensive clinical studies are still needed to thoroughly assess the effect of RTX against IgG4-RN and to gain insight into the timing and causes of relapse after treatment. Our unique case underlines the importance of a renal biopsy in undefined renal disease to understand the diagnosis and customize the treatment according to the patient’s needs.
References
Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061–7. https://doi.org/10.1002/art.34593.
Bledsoe JR, Della-Torre E, Rovati L, Deshpande V. IgG4-related disease: review of the histopathologic features, differential diagnosis, and therapeutic approach. APMIS. 2018;126(6):459–76. https://doi.org/10.1111/apm.12845.
Deshpande V. IgG4-related disease. Introduction Semin Diagn Pathol. 2012;29(4):175–6. https://doi.org/10.1053/j.semdp.2012.07.006.
Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas–an autonomous pancreatic disease? Am J Dig Dis. 1961;6:688–98.
Aoki S, Nakazawa T, Ohara H, et al. Immunohistochemical study of autoimmune pancreatitis using anti-IgG4 antibody and patients’ sera. Histopathology. 2005;47(2):147–58.
Deshpande V, Mino-Kenudson M, Brugge W, Lauwers GY. Autoimmune pancreatitis: more than just a pancreatic disease? A contemporary review of its pathology. Arch Pathol Lab Med. 2005;129(9):1148–54.
Igarashi H, Ito T, Oono T, Nakamura T, et al. Relationship between pancreatic and/or extrapancreatic lesions and serum IgG and IgG4 levels in IgG4-related diseases. J Dig Dis. 2012;13(5):274–9. https://doi.org/10.1111/j.1751-2980.2012.00583.x.
Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22(7):1343–52. https://doi.org/10.1681/ASN.2011010062.
Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitialnephritis. Kidney Int. 2010;78(10):1016–23. https://doi.org/10.1038/ki.2010.271.
Cortazar FB, Stone JH. IgG4-related disease and the kidney. Nat Rev Nephrol. 2015;11(10):599–609. https://doi.org/10.1038/nrneph.2015.95.
Pradhan D, Pattnaik N, Silowash R, Mohanty SK. IgG4-related kidney disease–A review. Pathol Res Pract. 2015;211(10):707–11. https://doi.org/10.1016/j.prp.2015.03.004.
Zhang P, Cornell LD. IgG4-related tubulointerstitial nephritis. Adv Chronic Kidney Dis. 2017;24(2):94–100. https://doi.org/10.1053/j.ackd.2016.12.001.
Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–92. https://doi.org/10.1038/modpathol.2012.72.
Salvadori M, Tsalouchos A. Immunoglobulin G4-related kidney diseases: an updated review. Word J Nephrol. 2018;7(19):29–40.
Hara S, Kawano M, Mizushima I, et al. Distribution and components of interstitial inflammation and fibrosis in IgG4-related kidney disease: analysis of autopsy specimens. Hum Pathol. 2016;55:164–73. https://doi.org/10.1016/j.humpath.2016.05.010.
Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011. https://doi.org/10.1681/ASN.2011010062.
Kawano M, Saeki T. IgG4-related kidney disease–an update. Curr Op in Nephrol Hypertens. 2015;24(2):193–201. https://doi.org/10.1097/MNH.0000000000000102.
Vaglio A, Salvarani C, Buzio C. Retroperitoneal fibrosis. Lancet. 2006;367(9506):241–51.
Quattrocchio G, Roccatello D. IgG4-related nephropathy. J Nephrol. 2016;29(4):487–93. https://doi.org/10.1007/s40620-016-0279-4.
Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466–75. https://doi.org/10.1002/art.39205.
Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15(5):615–26. https://doi.org/10.1007/s10157-011-0521-2.
Kim B, Kim JH, Byun JH, et al. IgG4-related kidney disease: MRI findings with emphasis on the usefulness of diffusion-weighted imaging. Eur J Radiol. 2014;83(7):1057–62. https://doi.org/10.1016/j.ejrad.2014.03.033.
Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–51. https://doi.org/10.1056/NEJMra1104650.
Kamisawa T, Okazaki K. Diagnosis and Treatment of IgG4-Related Disease. Curr Top Microbiol Immunol. 2017;401:19–33. https://doi.org/10.1007/82_2016_36.
Khosroshahi A, Wallace ZS. Crowe JL et al International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015;67(7):1688–99. https://doi.org/10.1002/art.39132.
Masaki Y, Shimizu H, Sato Nakamura T, et al. IgG4-related disease: diagnostic methods and therapeutic strategies in Japan. J Clin Exp Hematop. 2014;54(2):95–101.
Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–7.
Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut. 2007;56:1650–2.
Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15.
Raina A, Yadav D, Krasinskas AM, et al. Evaluation and management of autoimmune pancreatitis: experience at a large US center. Am J Gastroenterol. 2009;104:2295–306.
Kamisawa T, Okazaki K, Kawa S, et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013. III. Treatment and prognosis of autoimmune pancreatitis. J Gastroenterol. 2014;49:961–70.
Mizushima I, Yamada K, Fujii H, et al. Clinical and histological changes associated with corticosteroid therapy in IgG4-related tubulointerstitial nephritis. Mod Rheumatol. 2012;22(6):859–70. https://doi.org/10.1007/s10165-011-0589-2.
Mise N, Tomizawa Y, Fujii A, Yamaguchi Y, Sugimoto T. A case of tubulointerstitial nephritis in IgG4-related systemic disease with markedly enlarged kidneys. NDT Plus. 2009;2(3):233–5. https://doi.org/10.1093/ndtplus/sfp023.
Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171–7. https://doi.org/10.1136/annrheumdis-2014-206605.
Yamamoto M, Awakawa T, Takahashi H. Is rituximab effective for IgG4-related disease in the long term? Experience of cases treated with rituximab for 4 years. Ann Rheum Dis. 2015;74(8):e46.
Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91(1):57–66. https://doi.org/10.1097/MD.0b013e3182431ef6.
Roccatello D, Sciascia S, Rossi D, Alpa M, Naretto C, Baldovino S. Intensive short-term treatment with rituximab, cyclophosphamide and methylprednisolone pulses induces remission in severe cases of SLE with nephritis and avoids further immunosuppressive maintenance therapy. Nephrol Dial Transplant. 2011;26(12):3987–92. https://doi.org/10.1093/ndt/gfr109.
Hart PA, Topazian MD, Witzig TE, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62:1607–15.
Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients with IgG4-related disease. Medicine (Baltimore). 2012;91:57–66.
Soh SB, Pham A, O’Hehir RE, et al. Novel use of rituximab in a case of Riedel’s thyroiditis refractory to glucocorticoids and tamoxifen. J Clin Endocrinol Metab. 2013;98:3543–9.
Funding
The authors have no relevant financial or non-financial interests to disclose, and no funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
The patient has consented to the submission of the case report to the journal.
Ethical approval
Approved by the Local Ethical Committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Spatola, L., Ravera, F., Sghirlanzoni, M.C. et al. An enigmatic case of IgG4-related nephropathy and an updated review of the literature. Clin Exp Med 21, 493–500 (2021). https://doi.org/10.1007/s10238-021-00696-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-021-00696-x