
Abstract
Understanding the formation mechanisms of polymeric silicates is essential to the study of microbiology and biogeochemistry. It has implications for the growth of diatoms and dinoflagellates and studying the processes that control the dissolution, precipitation, and biological uptake of different silicates species can provide an understanding of the occurrence of toxic blooms. This study examines the seasonal distribution of monomeric and polymeric silicates in the brackish and freshwater lakes of Japan. Inductively coupled plasma atomic emission spectroscopy was used to detect and quantify total dissolved silicates (TSi) and the spectrophotometric molybdenum blue method was used to detect molybdate reactive silicates (monomers to tetramers). The difference between the concentrations obtained via these two methods was used to determine the concentrations of polymeric silicates. Polymeric silicates were detected in anoxic-reducing pore waters from sediments of the freshwater Lake Biwa and Lake Kawaguchi in Japan, with a maximum concentration of 0.42 mmol L−1. Polymeric silicate was continuously detected as long as the lake bottom environments remained under anoxic-reducing conditions. It provides insights on the formation mechanisms of polymeric silicates in freshwater lakes. The polymerization of silicates is understood to occur during the adsorption reaction between monomeric silicates and Fe(OH)3 precipitate. Furthermore, this polymerization is deemed to be a dehydration condensation reaction because the silicates adsorbed on Fe(OH)3 precipitate are situated at short distances from each other. In the anoxic-reducing environments, these monomeric and polymeric silicates are released from ferric hydroxide (Fe(OH)3) precipitate by reacting with hydrogen sulfide.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dissolved silicate is an essential nutrient for aquatic organisms, and it is mainly used by diatoms to form their skeletons (Perry and Keeling-Tucker 2000; Yee et al. 2003; Lacombe et al. 2007). Diatoms account for approximately 25% of the global net primary production of aquatic organisms (Willén 1991) and interfere with the growth of dinoflagellates which are among the leading causes of red tides (Furumai 2012).
Dissolved silicate is supplied to lakes and oceans through rivers and groundwater (Ning 2002; Lacombe et al. 2007; Cornelis et al. 2011). Soluble silicate is usually in the form of a monomer (H4SiO4). The average concentrations of dissolved silicate are 0.2 mmol L−1 in freshwaters (Aston 1983; Willén 1991) and 0.1 mmol L−1 in oceans (Tréguer et al. 1995).
Dissolved silicate is removed from the water column via two pathways: biological uptake, such as that which occurs during excessive growth of diatoms (Schelske and Stoermer 1971; Li et al. 2006); and precipitation, caused by the adsorption of dissolved silicate onto Fe(OH)3 precipitate (Tallberg 2000). These precipitates sink to the bottoms of lakes and oceans and are buried in the sediment for long periods, as long as the environment remains oxidative. Eventually, the dissolved silicate in a lake becomes depleted (Fischer and Knoll 2009).
When the concentration of dissolved monomeric silicate exceeds 1.4 mmol L−1 in 15 °C water, the silicates are converted to various polymeric silicate species (Zuhl and Amjad 2013). Some researchers have mentioned the presence of polymeric silicate species in natural waters with low molecular weights, ranging from silicate dimers to pentamers, at concentrations below their solubilities (George et al. 2000; Tanaka et al. 2013). These silicate species can be measured using the molybdenum blue method (O'Connor 1961).
Diatoms can biologically utilize dissolved silicates ranging from monomers to linear tetramers (Tanaka et al. 2013). The growth of diatoms is depressed by a lack of these light silicates and an increase in polymeric silicates with high molecular weights. A lack of diatom growth results in the rapid growth of harmful phytoplankton (Schelske and Stoermer 1971; Conley et al. 1993), such as dinoflagellates that produce neurotoxins (Wang 2008). Therefore, a study on the formation mechanisms of polymeric silicates is essential from the perspectives of microbiology and biogeochemistry.
Gas–liquid chromatography (Tarutani 1989) and nuclear magnetic resonance spectra (Sjöberg 1996) are applied as qualitative and quantitative methods for analyzing polymeric silicates. However, the most convenient analytical method is to combine inductively coupled plasma atomic emission spectroscopy (ICP-AES) and the spectrophotometric molybdenum blue method. According to O'Connor (1961), dissolved silicates that are monomers to linear pentamers are measurable within 5 min of the reaction time using the molybdenum blue method. The ICP-AES can measure TSi irrespective of the chemical species in the solutions (Tallberg 2000). Therefore, the concentration of polymeric silicates larger than pentamers can be calculated as the difference between the analytical results of ICP-AES and those of the molybdenum blue method.
Isshiki et al. (1991) analyzed seawater using this method and reported that the concentration of TSi increased with water depth; however, they could not observe a difference between ICP-AES results and those of the molybdenum blue method. They, therefore, concluded that polymeric silicate did not exist in seawater.
However, polymeric silicates have been found to occur seasonally in the pore water of freshwater lakes such as Lakes Biwa and Kawaguchi in Japan (Park and Sugiyama 2018). Although the TSi concentrations were lower than the solubility of monomeric silicate, polymeric silicates were detected in the pore water. In contrast, polymeric silicates were not detected in brackish lake waters since the analytical results of ICP-AES and the molybdenum blue method were precisely the same for all brackish lakes such as Lakes Nakaumi, Suigetsu, and Suga in Japan. In freshwater lakes, the lake bottom environment changes to an anoxic-reducing environment during summer, causing a rapid increase in the concentrations of TSi and H2S + HS− in the pore water. This phenomenon indicates that the elution mechanism of silicates in freshwater lakes may be similar to that of phosphates in anoxic-reducing environments (Valdes et al. 2002). Although Swedlund and Webster (1999) reported that the eluted silicate was adsorbed onto the surface of Fe(OH)3 to form a polymer, there has been no report on the chemical species of dissolved silicates in anoxic-reducing lake sediments. Furthermore, the adsorption and desorption processes of dissolved silicates and the stability of polymeric silicates remain poorly understood. In this study, we discuss the seasonal and vertical distributions of monomeric and polymeric silicates in the sediment of some brackish and freshwater lakes of Japan and clarify the formation mechanism of polymeric silicate by investigating polymerization sites and timing that occurs during the adsorption reaction between dissolved silicates and Fe(OH)3.
Materials and methods
Study areas
As the representative of brackish lakes, we selected Lakes Nakaumi, Suigetsu, and Suga in Japan. These lakes have sufficiently abundant dissolved salts to classify them as brackish lakes. The salinities in the hypolimnion layers are all above 10 psu (g kg−1), at 30, 12, and 11 psu for Lakes Nakaumi, Suigetsu, and Suga, respectively.
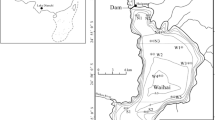
Previous studies have reported that these lakes are eutrophic (Maekawa et al. 1982; Kusunoki and Sakata 2018). We observed that anoxic and reducing conditions were reinforced during the summer season at the sampling points, located at 35.43°N, 144.27°E in Lake Nakaumi, 35.58°N, 135.88°E in Lake Suigetsu, and 35.58°N, 135.90°E in Lake Suga (Fig. 1). The water depths at the sampling points in Lakes Nakumi, Suigetsu, and Suga were 15, 33, and 12 m, respectively.

Sampling points in each lake (Lake Nakaumi, 35.43°N 144.27°E; Lake Suigetsu, 35.58°N 135.88°E; Lake Suga, 35.58°N 135.90°E; Lake Biwa, 35.00°N 135.57°E; Lake Kawaguchi, 35.51°N 138.73°E)
As the representative of freshwater lakes, we selected Lakes Biwa and Kawaguchi in Japan. Lake Biwa is the largest lake in Japan. It is situated in the western part of Japan (Fig. 1, 35.52°N, 136.20°E; Tanaka 1992) and can be divided into the northern and southern basins (Pollingher 1990). The southern basin is eutrophic and has an average depth of 5 m, with algal blooms occurring periodically in summer (Nalewajko and Murphy 2001). In this study, water and sediment samples were collected from February to December 2017 from an area (Fig. 1, 35.00°N, 135.57°E), in the south-eastern part of the southern basin, which was dredged to provide sediments and soils for the construction of an artificial island alongside it (Terashima and Ueda 1982). The samples were collected from a maximum depth of 13 m below the surface of the lake over an area of 0.25 km2. An anoxic hypolimnion typically appears in this area for several months of the year due to the thermal stratification of lake water during summer (Kawashima et al. 1985).
Lake Kawaguchi is located in the central part of Japan (Kyotani et al. 2005; Yamamoto et al. 2017). This lake is eutrophic, can be divided into the western, eastern, and Funatsu (south-eastern) basins and has a maximum water depth of 16.1 m (Nagasaka et al. 2002). Samples were collected at 35.51°N, 138.73°E from a depth of 11.3 m in the western basin (Fig. 1), where dissolved oxygen typically undergoes seasonal depletion in the hypolimnion layer, similar to the dredged area of Lake Biwa.
Sampling and pretreatment
Water depth, electrical conductivity, water temperature, dissolved oxygen (DO), and chlorophyll-a were measured using a multi water-quality profiler (Rinko-Profiler ASTD 102, JFE Advantech Co., Ltd., Japan). All water samples were collected using a water sampler (Niskin-X sampling bottles, General Oceanics, Inc., USA) and filtered using 0.45-μm polyvinylidene difluoride (PVDF) filters (Millex-HV syringe filter, Merck Millipore Ltd., Germany) within 2 h of collection. Ultrapure nitric acid (HNO3, 60%, Kanto Kagaku, Japan) was added to all filtrates to adjust the concentration of HNO3 to 0.02 mol L−1 and prevent the formation of Fe(OH)3.
Sediment samples were collected using a core sampler (HR-type core sampler, Rigo, Co., Inc., Japan). Pore water samples were filtered through a 0.45-μm PVDF filter, and HNO3 was added to the filtrate as described above.
Reagents
Highly purified Milli-Q water (Milli-Q Water Purification System ZD21-100sp, Millipore, Japan) was used in all laboratory experiments. Deoxygenated water was prepared by bubbling 99.99% nitrogen gas into 1000 mL of Milli-Q water for 1 h before use. In laboratory experiments, the pH of each sample solution was adjusted to 7 using dilute HCl or NaOH solutions.
Silicon standard solution (1000 mg-Si L−1, Na2SiO3 in 0.2 mol L−1 Na2CO3, Wako Pure Chemical Industries, Ltd., Osaka, Japan) was used to analyze silicates. Moreover, disodium molybdate (VI) dihydrate (Na2MoO4∙2H2O, Wako Pure Chemical Industries, Ltd., Osaka, Japan) was used as a coloring reagent. L-( +)-tartaric acid (Nakarai Tesque, INC., Kyoto, Japan) was used to remove the interference of phosphate. L-( +)-ascorbic acid (Wako Pure Chemical Industries, Ltd., Osaka, Japan) was used as a reducing agent for yellow silico-molybdate complexes.
The dissolved iron (DFe; Fe3+) solution was prepared using an iron standard solution (1000 mg-Fe L−1, Fe(NO3)3 in 0.275 mol L−1 HNO3 (Nakarai Tesque, INC., Kyoto, Japan)). The Na2S solution was prepared by dissolving sodium sulfide nonahydrate (Na2S·9H2O, Wako Pure Chemical Industries, Ltd., Osaka, Japan) in deoxygenated Milli-Q water, prepared as mentioned above.
Determination of dissolved silicate, dissolved iron, and total dissolved hydrogen sulfide
A spectrophotometer (Autoanalyzer, UV–VIS Spectrophotometer II, Bran Luebbe, Japan) and an inductively coupled plasma atomic emission spectrometer (ICP-AES; Optima 5000Z, Perkin-Elmer, Japan) were used to measure the concentration of dissolved silicate and distinguish the silicate species. Spectrophotometric determination of dissolved silicate was carried out using the molybdenum blue method at 660 nm (Hansen and Koroleff 1999).
In this study, the dissolved silicate concentrations measured using a spectrophotometer and ICP-AES are expressed as molybdate reactive silicates [MSi: silicates ranging from monomer to linear pentamer species (O’Connor 1961)] and TSi, respectively. The concentration of dissolved polymeric silicates (PSi: polymerized silicate species larger than pentamers) was calculated by subtracting MSi concentrations from TSi concentrations.
Dissolved iron and total hydrogen sulfide concentrations were analyzed using ICP-AES and spectrophotometry, respectively. Spectrophotometric determination of total hydrogen sulfide was carried out using the methylene blue method at 667 nm (Sugahara et al. 2010).
Laboratory experiments
Adsorption of dissolved silicate onto ferric hydroxide
In the adsorption reaction of dissolved silicates onto Fe(OH)3, the polymerization ratio of silicates was examined for 120 days. A mixed solution was prepared, containing 0.7 mmol L−1 of MSi and 0.1 mmol L−1 of DFe. To confirm the polymeric silicate production by adsorption, the molar concentration ratio of Si: Fe in the mixed solution was set to 7:1. The pH value of this solution was adjusted to 7.0 using the minimum required volume of concentrated NaOH or HCl solutions to form Fe(OH)3 precipitates. This solution was stored for 120 days at ambient temperature.
A 10 mL quantity of the mixed solution was collected on particular days, and then immediately filtered using a Nucleopore filter (pore size: 0.40 μm, diameter: 47 mm, Whatman). The MSi and TSi in the filtrate were then measured. The precipitate collected on the filter was reacted with 10 mL of 0.02 mol L−1 HNO3 solution for one day until it dissolved into the solution. The MSi and TSi concentrations in this solution were measured. PSi concentrations were calculated from these MSi and TSi concentrations.
Stability of PSi
The experiments were performed in the order shown in Fig. 2. The Fe(OH)3 was precipitated in a mixed solution (1.0 mmol L−1 of MSi, 0.1 mmol L−1 of DFe) by adjusting the pH of the solution to 7.0. After 6 months, this solution was divided into four fractions. Each fraction was then filtered through 0.40-μm Nucleopore filters. The MSi and TSi concentrations in each filtrate were measured using the molybdenum blue method and ICP-AES, respectively.

Schematic experimental process to evaluate PSi stability. The 0.4-µm nucleopore filter was used to separate the silicates adsorbed on Fe(OH)3 precipitate from the initial solution (the mixture of MSi and DFe). Moreover, the 0.2-µm nucleopore filter was used to remove FeS precipitate, including the colloidal FeS completely
The precipitates were treated using the procedure shown in Fig. 2. To quantify the relative proportions of MSi and PSi under oxic and anoxic conditions, we divided the experimental conditions into four fractions, such as HNO3 + Aeration, HNO3 + Anaeration, Na2S + Aeration, and Na2S + Anaeration.
The precipitate of the first fraction was added to 100 mL of 0.02 mol L−1 HNO3 (pH 2.20 ± 0.03) to dissolve the Fe(OH)3 precipitate. Prior to this procedure, the HNO3 solution was well aerated by bubbling atmospheric air filtered with air filters (CCF-050-D1B and CCF-010-D1B, Capsule filter, pore size: 0.5 and 0.1 μm, Advantec, Japan), and the DO concentration was approximately 8.9 mg-O2 L−1. This solution was re-filtered with a Nucleopore filter (pore size: 0.20 μm, diameter: 25 mm, Whatman). This re-filtrate was continuously monitored for changes in the concentrations of MSi, PSi, TSi, DO, and total dissolved hydrogen sulfide (H2S + HS−) for 14 days under ambient air conditions and room temperature. This experimental condition is hereafter referred to as “HNO3 + Aeration”.
The second fraction was also dissolved with 100 mL of 0.02 mol L−1 HNO3 (pH 2.20 ± 0.03). However, this HNO3 solution was first deoxygenated by bubbling with nitrogen gas. This solution was also re-filtered and then stored in a polyethylene bottle sealed with plastic film. This bottle was put into a polyethylene bag together with an open bottle containing Na2SO3 solution, as an oxygen-adsorbing agent, to protect from the invasion of atmospheric oxygen. This experimental condition is hereafter referred to as “HNO3 + Anaeration”. The filtrate was continuously monitored in a manner similar to that for the first fraction.
The precipitates from the third and fourth fractions were reacted with 100 mL of 0.3 mmol L−1 Na2S solution (pH 7.10 ± 0.05) for one day. The Fe(OH)3 precipitate was converted to ferrous sulfide precipitate. Here, the Na2S solution was prepared using deoxygenated Mili-Q water. Each solution was filtered again using a 0.20-μm Nucleopore filter to obtain the re-filtrate. These solutions were stored under aerobic and anaerobic conditions for 14 days, respectively. They were also continuously monitored, as in the case of the first fraction. These experimental conditions are hereafter referred to as “Na2S + Aeration” and “Na2S + Anaeration”, respectively.
Results and discussion
Distribution of silicate species in brackish lakes
Figures 3 and 4 show MSi and TSi concentrations in the pore waters of each brackish lake. In Lakes Nakaumi, Suigetsu, and Suga, MSi concentrations coincided with TSi concentrations in the pore waters. However, we noted the absence of PSi in the brackish lakes, even though the TSi concentration in the pore water of Lake Nakaumi was five times the solubility of MSi. In pore water of brackish lakes, the concentration of dissolved silicates exceeded by the solubility of MSi is attributed to the reduction of Fe(OH)3 by H2S in the anoxic-reducing environment; the subsequent reactions result in the formation of ferrous sulfide (FeS) and the dissolution of silicates that had been adsorbed onto Fe(OH)3. The same situation has been reported for Lake Nakaumi in our previous work (Park et al. 2020). Even from a thermodynamic point of view, this hypothesis is sufficiently valid. Fukusawa et al. (1995) have already shown that the iron in a brackish lake such as Lake Suigetsu is directly reduced by H2S. They suggest that the iron reduction process depends on the lake water characteristics. Furthermore, Lehtoranta et al. (2009) reported that the eutrophication of lakes had accelerated the conversion of the dominant species from iron-reducing bacteria to sulfate-reducing bacteria. This change in the biogeochemical characteristics of the sediments accelerates the production of H2S, which then directly reduces Fe(OH)3.

Vertical profiles of TSi (□), MSi (〇), and PSi (▲) in the pore water of Lake Nakaumi (August 21, 2017). No significant difference was observed between TSi and MSi concentrations

Vertical profiles of TSi (□), MSi (〇), and PSi (▲) in the pore waters of Lakes Suigetsu (left) and Suga (right) on May 20, 2017
Distribution of silicate species in freshwater lakes
Figure 5 shows the vertical distribution of DO in the water columns of Lakes Biwa (September 21, 2017) and Kawaguchi (September 30, 2017). Figures 6 and 7 show the vertical distributions of DFe, H2S + HS−, TSi, MSi, and PSi concentrations in pore waters of the freshwater lakes. In both lakes, the DO was depleted in the pore water and hypolimnion layer (Fig. 5). The TSi concentration in the pore water was slightly higher in Lake Biwa than in Lake Kawaguchi, but the difference was not significant. The PSi concentrations were 0–0.42 mmol L−1 in Lake Biwa and 0–0.23 mmol L−1 in Lake Kawaguchi (Fig. 7). The concentrations of DFe and H2S + HS− in the pore waters were higher in Lake Biwa compared to that in Lake Kawaguchi (Fig. 6).

Vertical profiles of DO in Lake Biwa (●, September 21, 2017) and Lake Kawaguchi (〇, September 30, 2017)

Vertical profiles of DFe and H2S + HS− in the pore waters of Lake Biwa (●, September 21, 2017) and Lake Kawaguchi (〇, September 30, 2017)

Vertical profiles of TSi (□), MSi (〇), and PSi (▲) in the pore waters of Lake Biwa (left; September 21, 2017) and Lake Kawaguchi (right; September 30, 2017)
Figure 8 shows the variation of DO concentration at a depth of 12 m (just above the lake bottom) in Lake Biwa from February 14 to December 15, 2017. From February to April and from October to December, the DO was high in the hypolimnion layer. However, from mid-May to mid-September, the environment of the bottom layer was anoxic and reducing (Fig. 8), and concentrations of DFe, H2S + HS−, and TSi increased (Fig. 9). The maximum PSi concentration in the pore water was 0.40 mmol L−1 at a depth of 5 cm in September 2017 and it accounted for approximately 33% of the TSi.

Annual change in DO concentration in the hypolimnion layer (water depth: 12 m) of Lake Biwa

Vertical profiles of DFe (■), H2S + HS− (△), TSi (□), MSi (〇), and PSi (▲) in the pore water of Lake Biwa from March 14 to December 15, 2017
The highest concentration of dissolved silicate was reported for ground water (1.0 mmol L−1; Krauskopf and Bird 1995) and it has been suggested that dissolved silicate exists in a polymeric form because, at this concentration, it is close to the monomeric silicate solubility (1.4 mmol L−1).
Van der Weijden (2007) stated that this highly concentrated dissolved silicate, eluted from the lake sediments, likely originated from biomaterials such as diatom debris. However, Lehtimäki et al. (2016) reported that the silicates eluted from sediments in an anoxic-reducing freshwater lake were derived from inorganic oxides such as metal oxides, and not biogenic silica.
Adsorption of dissolved silicate onto ferric hydroxide precipitate and its polymerization
Figure 10 shows the changes in the adsorption ratio of silicate onto Fe(OH)3 over 120 days. In this experiment, the fractions of PSi and MSi in the Fe(OH)3 precipitate were determined after the dissolution of the Fe(OH)3 precipitate in a 0.02 mol L−1 HNO3 solution, as described in the method section. The sum of TSi (PSi and MSi) concentrations in the filtrate and the precipitate coincided with the initial concentration of silicate detected at the beginning of the experiment. Only MSi was identified in the filtrate, while PSi was not detected during the entire experimental period.

Formation of polymeric silicate in the adsorption reaction of silicate onto ferric hydroxide, over 120 days. Only the concentrations of TSi (□), MSi (〇), and PSi (▲) in the precipitate are presented
The adsorption ratio of silicate continuously increased over time. The adsorption ratio was 45% on day 0, and then increased to 68% over the following 120 days. The concentration of MSi in the precipitate was 0.32 mmol L−1 on day 0 and had decreased to 0.21 mmol L−1 by the 40th day. On the 120th day, MSi in the precipitate had decreased to 0.20 mmol L−1. By contrast, the PSi concentration showed a value close to 0.00 mmol L−1 on the initial day but increased to 0.21 mmol L−1 on the 40th day and 0.29 mmol L−1 on the 120th day. The ratio of PSi to TSi concentrations in the precipitate increased from 0% on day 0 to 50% on the 40th day and 60% on the 120th day (Fig. 10). These results show that silicates were transferred from the aqueous solution to the precipitate via adsorption in the solution. After adsorption, MSi on Fe(OH)3 was polymerized to PSi.
It is noteworthy that PSi was only detected in the precipitate and that the initial MSi in the mixed solution was 0.7 mmol L−1, which was less than half of the solubility of MSi. Therefore, the adsorption of dissolved silicates is inferred as an essential process for the polymerization of silicates.
Stability of PSi
The annual change in the distribution profile of PSi and TSi in Lake Biwa (Fig. 9) shows that TSi concentrations were lower than 1.4 mmol L−1, but PSi concentrations were stable in the lake bottom throughout the anoxic-reducing period. Thus, it was not clear how the PSi could dissolve in the pore water for an extended period under such conditions.
Figure 11 presents the stability of PSi eluted from Fe(OH)3 precipitates in different conditions. A decrease in PSi concentration was observed only in aerobic conditions, namely the HNO3 + Aeration and Na2S + Aeration conditions. Furthermore, PSi concentration under HNO3 + Aeration was rapidly depleted, only two days after aeration. With Na2S + Aeration, PSi concentration decreased from 0.44 mmol L−1 on the 1st day to 0.34 mmol L−1 on the 3rd day (Fig. 11), when H2S + HS− decreased to 0 mmol L−1 (Fig. 12).

Stability of PSi under each set of conditions, namely HNO3 + Aeration (〇), HNO3 + Anaeration (□), Na2S + Aeration (●), and Na2S + Anaeration (▲), over 14 days

Changes in the concentration of H2S + HS− and DO under each set of conditions, over two weeks. □: DO in HNO3 + Aeration, ●: DO in Na2S + Aeration, △: H2S + HS− in Na2S + Aeration, and ▲: H2S + HS− in Na2S + Anaeration. Note that concentrations of DO in HNO3 + Anaeration, DO in Na2S + Anaeration, and H2S + HS− in HNO3 + Aeration/Anaeration have been omitted because these values were 0 mg-O2 L−1 (DO) or 0 mmol L−1 (H2S + HS−) throughout the experimental period
On the other hand, in anaerobic conditions (anoxic-reducing conditions, HNO3/Na2S + Anaeration), the initial PSi concentration was maintained for 14 days of the experiment, regardless of the chemical composition of the solution. Therefore, these results suggest that PSi is stabilized under anaerobic conditions. In other words, PSi is generally only observed in anaerobic water. Unfortunately, the precise mechanism by which PSi is stabilized under anaerobic conditions is unknown at present. We will study this mechanism in detail in the future.
Conclusion
Polymerization of dissolved silicate was found to occur after its adsorption onto Fe(OH)3 in an oxidative environment, even if the TSi concentration was lower than 1.4 mmol L−1, the solubility of monomeric silicate. The PSi was only detected in the pore water of freshwater lakes where H2S was generated in anoxic-reducing conditions. When H2S reacts with Fe(OH)3 to form FeS, MSi and PSi in the hydroxide are released into the pore water. Moreover, this PSi dissolves steadily in an anoxic-reducing environment.
Data availability
The data that support the findings of this study are available from the corresponding author, JY Park, upon reasonable request.
References
Aston S (1983) Natural water and atmospheric chemistry of silicon. In: Aston S (ed) Silicon geochemistry and biogeochemistry. Academic Press, Massachusetts, US, pp 77–100
Conley D, Schelske C, Stoermer E (1993) Modification of the biogeochemical cycle of silica with eutrophication. Mar Ecol Prog Ser 101:179–192. https://doi.org/10.3354/meps101179
Cornelis JT, Delvaux B, Georg RB, Lucas Y, Ranger J, Opfergelt S (2011) Tracing the origin of dissolved silicon transferred from various soil-plant systems towards rivers: a review. Biogeosciences 8:89–112. https://doi.org/10.5194/bg-8-89-2011
Fischer WW, Knoll AH (2009) An iron shuttle for deepwater silica in Late Archean and early Paleoproterozoic iron formation. Geol Soc Am Bull 121:222–235. https://doi.org/10.1130/B26328.1
Fukusawa H, Koizumi I, Okamura M, Yasuda Y (1995) Last 2,000 year records of eolian dust concentration sea-level and precipitation changes in fine-grained sediment of Lake Suigetsu, Central Japan (in Japanese). Chigaku Zasshi 104:69–81. https://doi.org/10.5026/jgeography.104.69
Furumai H (2012) Monitoring of silicic acid and research implication (in Japanese). In: Furumai H, Sato K, Yamamoto K (eds) Silicic acid, its source and transportation (in Japanese). Gihodo Shuppan, Tokyo, Japan. 165–176
George S, Steinberg SM, Hodge V (2000) The concentration, apparent molecular weight and chemical reactivity of silica from groundwater in Southern Nevada. Chemosphere 40:57–63. https://doi.org/10.1016/s0045-6535(99)00240-4
Hansen HP, Koroleff F (1999) Determination of nutrients. In: Grasshoff K, Kremling K, Ehrhardt M (eds) Methods of seawater analysis, 3rd edn. Wiley, pp 159–228
Isshiki K, Sohrin Y, Nakayama E (1991) Form of dissolved silicon in seawater. Mar Chem 32:1–8. https://doi.org/10.1016/0304-4203(91)90021-N
Kawashima M, Hori T, Koyama M, Takamatsu T (1985) Redox cycle of manganese and iron and the circulation of phosphorus in a dredged area of the Southern lake. In: Takamatsu T (ed), Limnological and environmental studies of elements in the sediment of Lake Biwa. The National Institute for Environmental Studies, Japan, 47–62
Krauskopf K, Bird D (1995) Sedimentation and Diagenesis: Inorganic Geochemistry. In: Krauskopf K, Bird D (eds) Introduction to Geochemistry, 3rd edn. McGraw-Hill, New York, US, pp 356–384
Kusunoki K, Sakata M (2018) Analysis of historical trend of eutrophication in Lake Nakaumi, Japan, using concentrations of several index elements in sediment cores (in Japanese). Mizu Kankyo Gakkaishi 41:151–157. https://doi.org/10.2965/jswe.41.151
Kyotani T, Koshimizu S, Kobayashi H (2005) Short-term cycle of eolian dust (Kosa) recorded in Lake Kawaguchi sediments, central Japan. Atmos Environ 39:3335–3342. https://doi.org/10.1016/j.atmosenv.2005.01.026
Lacombe M, Garçon V, Comtat M, Oriol L, Sudre J, Thouron D, Le Bris N, Provost C (2007) Silicate determination in sea water Toward a reagentless electrochemical method. Mar Chem 106:489–497. https://doi.org/10.1016/j.marchem.2007.05.002
Lehtimäki M, Sinkko H, Tallberg P (2016) The role of oxygen conditions in the microbial dissolution of biogenic silica under brackish conditions. Biogeochemistry 129:355–371. https://doi.org/10.1007/s10533-016-0237
Lehtoranta J, Ekholm P, Pitkänen H (2009) Coastal eutrophication thresholds: a matter of sediment microbial processes. Ambio 38:303–308. https://doi.org/10.1579/09-A-656.1
Li X, Song J, Dai J, Yuan H, Li N, Li F, Sun S (2006) Biogenic silicate accumulation in sediments, Jiaozhou Bay. Chin J Oceanol Limnol 24:270–277. https://doi.org/10.1007/BF02842627
Maekawa K, Shirasaki K, Sawada T, Yamaguchi S, Utsunomiya T, Aoki K, Tagawa S, Isomatsu Y (1982) Study on the eutrophication of Lakes Mikata Goko: Horizontal distribution of water quality of the lakes (in Japanese). Annual Report of The Environmental Pollution Research Center of Fukui Prefecture, Japan, 181–185
Nagasaka M, Yoshizawa K, Ariizumi K, Hirabayashi K (2002) Temporal changes and vertical distribution of macrophytes in Lake Kawaguchi. Limnology 3:107–114. https://doi.org/10.1007/s102010200012
Nalewajko C, Murphy T (2001) Effects of temperature, and availability of nitrogen and phosphorus on the abundance of Anabaena and Microcystis in Lake Biwa, Japan: an experimental approach. Limnology 2:45–48. https://doi.org/10.1007/s102010170015
Ning R (2002) Discussion of silica speciation, fouling, control and maximum reduction. Desalination 151:67–73. https://doi.org/10.1016/S0011-9164(02)00973-6
O’Connor T (1961) The reaction rates of polysilicic acids with molybdic acid. J Phys Chem 65:1–5. https://doi.org/10.1021/j100819a001
Park JY, Sugahara S, Egawa M, Seike Y (2020) Mechanism of silicate elution by hydrogen sulfide from bottom sediment in a brackish lake. Limnology 21:197–205. https://doi.org/10.1007/s10201-019-00601-2
Park JY, Sugiyama M (2018) Formation and decomposition of polymeric silicate in pore water, 17th World Lake Conference proceedings, Lake Kasumigaura, Ibaraki, Japan, 1159–1161
Perry C, Keeling-Tucker T (2000) Biosilicification: the role of the organic matrix in structure control. J Biol Inorg Chem 5:537–550. https://doi.org/10.1007/s007750000130
Pollingher U (1990) Effects of latitude on phytoplankton composition and abundance in large lakes. In: Tilzer M, Serruya C (eds) Large lakes: ecological structure and function. Springer, Berlin, Germany, pp 368–402
Schelske C, Stoermer E (1971) Eutrophication, silica depletion, and predicted changes in algal quality in Lake Michigan. Science 173:423–424. https://doi.org/10.1126/science.173.3995.423
Sjöberg S (1996) Silica in aqueous environment. J Non-Cryst Solids 196:51–57. https://doi.org/10.1016/0022-3093(95)00562-5
Sugahara S, Yurimoto T, Ayukawa K, Kimoto K, Senga Y, Okumura M, Seike Y (2010) A simple in situ extraction method for dissolved sulfide in sandy mud sediments followed by spectrophotometric determination and its application to the bottom sediment at the Northeast of Ariake Bay (in Japanese). Bunseki Kagaku 59:1155–1161. https://doi.org/10.2116/bunsekikagaku.59.1155
Swedlund PJ, Webster JG (1999) Adsorption and polymerisation of silicic acid on ferrihydrite, and its effect on arsenic adsorption. Water Res 33:3413–3422. https://doi.org/10.1016/S0043-1354(99)00055-X
Tallberg P (2000) Silicon and its impacts on phosphorus in eutrophic freshwater lakes. Ph. D. thesis. Univ of Helsinki
Tanaka M (1992) The Lakes in Japan (in Japanese). Nagoya University, Japan
Tanaka M, Takahashi K, Nemoto M, Horimoto N (2013) Selectivity of silica species in ocean observed from seasonal and local changes. Spectrochim Acta, Part A 104:423–427. https://doi.org/10.1016/j.saa.2012.11.040
Tarutani T (1989) Polymerization of silicic acid a review. Anal Sci 5:245–252. https://doi.org/10.2116/analsci.5.245
Terashima A, Ueda T (1982) Effect of bottom dredging on some environmental factors and benthic animals in the South basin of Lake Biwa (in Japanese). Jpn J Limnol 43:81–87. https://doi.org/10.3739/rikusui.43.81
Tréguer P, Nelson D, Van Bennekom A, de Master D, Leynaert A, Quéguiner B (1995) The silica balance in the world ocean: a reestimate. Science 268:375–379. https://doi.org/10.1126/science.268.5209.375
Valdes LM, Price KS, Luther GW III (2002) Iron sulfur phosphorus cycling in the sediments of a shallow coastal bay: implications for sediment nutrient release and benthic macroalgal blooms. Limnol Oceanogr 47:1346–1354. https://doi.org/10.4319/lo.2002.47.5.1346
Wang D (2008) Neurotoxins from marine dinoflagellates: a brief review. Mar Drugs 6:349–371. https://doi.org/10.3390/md20080016
Van der Weijden C (2007) Silica I: silicon analytical, physical and terrestrial geochemistry, Ph. D. thesis. Utrecht University
Willén E (1991) Planktonic diatoms - an ecological review. Archiv für Hydrobiologie. Supplementband, Algological studies 62: 69–106. ISSN 0342–1120
Yamamoto S, Nakamura T, Uchiyama T (2017) Newly discovered lake bottom springs from Lake Kawaguchi, the Northern foot of Mount Fuji Japan. J Jap Assoc Hydrol Sci 47:49–59. https://doi.org/10.4145/jahs.47.49
Yee N, Phoenix V, Konhauser K, Benning L, Ferris G (2003) The effect of cyanobacteria on silica precipitation at neutral pH: implications for bacterial silicification in geothermal hot springs. Chem Geol 199:83–90. https://doi.org/10.1016/S0009-2541(03)00120-7
Zuhl RW, Amjad Z (2013) Solution chemistry impact on silica polymerization by inhibitors. In: Amjad Z (ed) Mineral Scales in Biological and Industrial Systems. CRC Press, Florida, US, pp 173–200
Acknowledgements
We thank the Center for Ecological Research, Kyoto University for the use of their research vessel "Hasu" and observation facilities. We thank Dr. Y. Goda, Dr. T. Akatsuka, and Prof. S. Nakano of the Center for Assistance for support during field work. This work was partly supported by JSPS KAKENHI vide Grant Number: 17K00518.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Yuko Sugiyama.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, J.Y., Sugiyama, M., Sugahara, S. et al. Detection of polymeric silicate in the pore water of freshwater lakes. Limnology 24, 171–179 (2023). https://doi.org/10.1007/s10201-023-00716-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10201-023-00716-7