
Abstract
Background
Despite previous therapeutic studies on autophagy in cancer, its role in the treatment of pancreatic ductal adenocarcinoma remains controversial, especially regarding its effect on chemotherapy, radiotherapy, and both combined. We focused on RUN domain Beclin-1 interacting and cysteine-rich-containing protein (Rubicon) to reveal its contribution to pancreatic ductal adenocarcinoma after chemoradiotherapy.
Methods
To evaluate the clinical significance of Rubicon, immunohistochemistry was performed, and Rubicon expression was analyzed across 81 specimens resected from patients with pancreatic ductal adenocarcinoma after neoadjuvant chemoradiotherapy. A gemcitabine-resistant pancreatic ductal adenocarcinoma cell line was established followed by Rubicon expression and autophagy flux estimation. Finally, gemcitabine sensitivity, invasion ability, and cell viability were evaluated using Rubicon-targeting small interfering RNA.
Results
Rubicon expression in resected pancreatic ductal adenocarcinoma samples after chemoradiotherapy revealed significantly worse overall survival and recurrence-free survival in the Rubicon-high expression group than in the Rubicon-low expression group (overall survival: median [years] 2.02 vs. 3.21, p = 0.0359; recurrence-free survival: median [years] 0.90 vs. 1.90, p = 0.0146). In vitro, gemcitabine-resistant pancreatic ductal adenocarcinoma cell lines exhibited higher Rubicon expression and lower autophagy flux than the parental cell line (p < 0.01). Transduction with small interfering RNA downregulated the expression without affecting gemcitabine sensitivity, but it reduced invasion ability and cell viability (p < 0.01) in the gemcitabine-resistant pancreatic ductal adenocarcinoma cell line.
Conclusions
High Rubicon expression is a significant, unfavorable prognostic factor in pancreatic ductal adenocarcinoma after neoadjuvant chemoradiotherapy. Downregulation of Rubicon expression improves invasion ability and cell viability in gemcitabine-resistant pancreatic ductal adenocarcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) develops from pancreatic duct epithelial cells and is a fatal neoplasm with a poor prognosis [1]. PDAC treatment necessitates a multidisciplinary approach including chemotherapy and radiotherapy. Neoadjuvant therapy plays an important role in PDAC treatment for reducing tumor size and controlling potential micrometastases [2]. Several studies have reported the benefit of neoadjuvant chemoradiotherapy for PDAC [3,4,5]. Despite these advancements in the treatment and management of PDAC, its 5-year survival rate remains at 9% [6, 7].
To improve the prognosis of PDAC, studies on autophagy as a potential therapeutic target have gained increasing attention. Autophagy is essential for cell development, differentiation, homeostasis, and survival. Autophagy maintains tissue and cell homeostasis by digesting damaged components, recycling materials, and producing energy; its dysregulation has been involved in the pathogenesis of a variety of human diseases, including cancer [8, 9]. Cancer cells survive cancer therapy by processing organelles and proteins damaged by chemotherapy and radiotherapy, employing autophagy. In this pathway, antimalarial drugs, such as chloroquine (CQ) and hydroxychloroquine (HCQ), inhibit the autophagosome–lysosome infusion, thereby suppressing autophagy [10]. Previous in vitro findings revealed that the combination therapy of gemcitabine (GEM) with CQ inhibits PDAC growth more effectively than GEM alone [11]. Moreover, a clinical study involving the addition of HCQ to preoperative GEM and nab-paclitaxel chemotherapy in patients with resectable PDAC showed that although the preoperative addition of HCQ improved pathological and biomarker (cancer antigen 19–9) response, it did not improve prognosis in terms of overall survival (OS) or recurrence-free survival (RFS) in PDAC [12]. On the contrary, anticancer drugs induce autophagy in PDAC cells, enhancing the cytotoxicity of anticancer drugs and contributing to cell death [13, 14]. Thus, the role of autophagy in cancer therapy for PDAC remains controversial, and whether it contributes to cancer survival or suppresses cancer progression during treatment remains unclear. Moreover, to the best of our knowledge, no studies until now have reported improved prognoses associated with these theories.
To evaluate autophagy in cancer therapy for PDAC, this study focused on RUN domain Beclin-1 interacting and cysteine-rich containing protein (Rubicon), an endogenous negative regulator of autophagy. Rubicon interacts with the UV radiation resistance-associated gene (UVRAG)-Beclin1-P13KC13 (class III phosphatidylinositol-3 kinase) complex, localizes at endosomes and lysosomes, and prevents proper autophagolytic breakdown, especially during the autophagosome–lysosome fusion and endocytic trafficking [15]. Previously, Rubicon was associated with lifestyle-related diseases, such as non-alcoholic fatty liver disease (NAFLD) and cholestasis, and also with aging [16,17,18]. In Hep G2 cells, Rubicon was found to be upregulated, thus decreasing autophagy flux by supplementation with fatty acid palmitate. This induced endoplasmic reticulum stress and lipid accumulation. Moreover, Rubicon was also upregulated in association with autophagy impairment in the livers of mice fed a high-fat diet. In humans, liver tissues obtained from patients with NAFLD have been shown to express significantly higher levels of Rubicon than those without steatosis [16].
Therefore, we aimed to examine the impact of neoadjuvant therapy on Rubicon in PDAC cells to investigate the role of autophagy in cancer therapy for PDAC. We evaluated Rubicon expression in PDAC after neoadjuvant therapy and its association with prognosis. In addition, we also investigated the expression of Rubicon, autophagy flux, and the effect of Rubicon suppression in a GEM-resistant (GR)-PDAC cell line that survived long-term exposure to GEM.
Methods
Clinical samples
We performed immunohistochemistry on samples from 81 Asian patients treated for PDAC with neoadjuvant chemoradiotherapy between March 2007 and May 2015 at Osaka University Hospital. The use of resected samples was approved by the Human Ethics Review Committee of the Graduate School of Medicine, Osaka University (Certificate Number 21264-2). Written informed consent was obtained from each patient. For neoadjuvant chemotherapy, GEM and oral fluoropyrimidine were typically used and radiotherapy was delivered to a total dose of 50.4 Gy. The clinicopathological features of the patients are shown in Supplementary Table 1.
Cell lines, culture, and materials
The human PDAC cell line MIAPaCa-2 was purchased from the Japan Cancer Research Resources Bank (Tokyo, Japan). Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, 100 mg/mL streptomycin, and 100 U/mL penicillin at 37 °C in a humidified incubator with 5% CO2.
Establishment of a gemcitabine-resistant MIAPaCa-2 clone (MIA-GR)
GR cells were generated by exposure to GEM for 2 months, as described previously [19, 20]. Parental MIAPaCa-2 cells were exposed to GEM at an initial concentration of 1 ng/ml. As cells adapted to GEM, the concentration was gradually increased finally reaching 20 ng/ml. Through this process, the GR PDAC cell line was established [21]. Three clones (MIA-GR1, 2, and 3) were used in these experiments.
Immunohistochemistry
Immunohistochemical studies of all specimens were performed to detect Rubicon expression, as described in previous studies [22, 23]. Briefly, formalin-fixed, paraffin-embedded tissues were deparaffinized, boiled for antigen retrieval, and incubated with a Rubicon-specific antibody (rabbit polyclonal, 21,444-1-AP, 1:200 dilution, Proteintech Group, Chicago, IL, USA) overnight at 4℃. Bound antibodies were detected using biotin-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA). We evaluated cytoplasmic Rubicon expression in the cancer lesions of PDAC specimens relative to Rubicon expression in the cytoplasm and membrane of the splenic red pulp, which was the positive control and divided into a strongly positive group (stronger staining intensity than that in the cytoplasm and membrane of splenic red pulp) and weakly positive group (similar or weaker staining intensity than that in the cytoplasm and membrane of splenic red pulp). This evaluation was independently performed by KG and SK, who were blinded to the clinical outcomes.
Western blotting
Western blotting (WB) was performed as described in a previous study [24]. Briefly, total protein was extracted from PDAC cell lines. Aliquots of total protein (10 μg) were electrophoresed on sodium dodecyl sulfate-polyacrylamide/Tris–HCl gels (Bio-Rad Laboratories, Hercules, CA, USA). The separated proteins were transferred to polyvinylidene difluoride membranes and incubated with primary antibodies overnight at 4 °C. The membranes were probed with the antibodies listed in the Supplementary Material.
Quantitative real-time polymerase chain reaction
We conducted a quantitative real-time polymerase chain reaction (qRT-PCR) as previously described [25]. qRT-PCR was then performed with gene-specific oligonucleotide primers (Supplementary Table 2) on an Applied Biosystems® ViiA™7 Real-Time PCR System (Thermo Fisher Scientific, MA, USA). Amplification products were quantified using THUNDERBIRD® SYBR qPCR Mix (TOYOBO, Osaka, Japan). Target gene expression levels were normalized to the expression level of beta-actin, which served as an internal control.
Downregulation of Rubicon expression by small interfering RNA
MIA-GR cell line was transfected with 5 nM small interfering RNA (siRNA) targeting Rubicon expression (Silencer Select Pre-designed siRNA, siRNA ID s18717 and s18718; Life Technologies, Carlsbad, CA, USA) using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The same method was applied with non-targeting siRNA (Silencer Select negative control #1 siRNA; Life Technologies) as the control.
Autophagic flux assay
Cells were treated with 125 nM of Bafilomycin (BioViotica, Dransfeld, Germany), in dimethyl sulfoxide 2 h before harvest and were subjected to WB. The autophagic flux index was calculated as the ratio of the expression level of microtubule-associated protein light chain 3 (LC3)-II in the presence of Bafilomycin to the expression level of LC3-II in the absence of Bafilomycin. The expression level of LC3-II was normalized to beta-actin as a loading control. The autophagic flux index of the control group in each experiment was normalized to 1 [26,27,28].
Growth inhibition assays with gemcitabine therapy
Growth inhibition was assessed using the MTT assay (Sigma-Aldrich, St. Louis, MO, USA), as described previously [29]. Each cell line was incubated for 24 h. Subsequently, cells were exposed to GEM for 72 h. Cell viability was evaluated by measuring absorbance in the MTT solution using a microplate reader. The results are expressed as the percentage of absorbance relative to that of untreated controls.
Cell viability assays
Tumor cell viability was assessed as previously described [30]. MIA-GR cell proliferation was determined using a Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Kumamoto, Japan). The cells were incubated for 24 h, and the CCK-8 solution was added to each well. The cells were incubated for 2 h, and the absorbance was measured using a microplate reader. The results are expressed as the percentage absorbance after 48 and 72 h of CCK-8 exposure relative to 0 h of exposure.
Invasion assay
The invasion assay was performed with invasion chambers loaded with Matrigel according to the manufacturer’s instructions as described previously [31]. Briefly, 5 × 104 cells were overlaid onto a Matrigel matrix on a membrane with 8‐mm‐diameter pores with or without Vorinostat or DHOB. After 48 h, the cells that had invaded the undersurface of the membrane were fixed with methanol and stained with thiazine and eosinate. Five microscopic fields were randomly selected for cell counting.
Statistical analyses
This information is listed in the Supplementary Material.
Results
Patients with PDAC after neoadjuvant therapy with Rubicon expression had a significantly worse prognosis
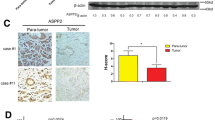
Rubicon expression was investigated using immunohistochemistry in PDAC specimens from curative resections after neoadjuvant chemoradiotherapy. Compared to spleen specimens (the positive control), PDAC cells displayed increased Rubicon staining in their cytoplasm. The 81 specimens were divided into two groups based on staining intensity compared with the cytoplasm and membrane of splenic red pulp: strongly Rubicon-positive (n = 33) and weakly Rubicon-positive (n = 48) groups (Fig. 1a–c). The clinicopathological characteristics of each group are summarized in Supplementary Table 3. Analyses of the relationship between Rubicon expression and clinicopathological features showed no significant associations among these factors, including pathological residual cancer, number of lymph node metastasis, and completion of adjuvant chemotherapy. The mean follow-up durations were 3.72 ± 2.99 years and 2.82 ± 3.00 years for patients with OS and RFS, respectively. The survival analysis revealed that patients with strong Rubicon expression had a significantly worse outcome than patients with weak Rubicon expression (median OS: 2.02 vs. 3.21, p = 0.0359; median RFS: 0.90 vs. 1.90, p = 0.0146) (Fig. 1d, e). In the univariate analysis of OS and RFS, two clinicopathological features were found to be significantly associated with OS and RFS: the completion of adjuvant therapy (OS: p = 0.0205, RFS: p = 0.0291) and Rubicon expression (OS: p = 0.0401, RFS: p = 0.0178). Moreover, in the multivariate analysis of these risk factors, the completion of adjuvant therapy (OS: p = 0.0113, RFS: 0.0171) and Rubicon expression (OS: p = 0.0217, RFS: 0.0105) were prognostic factors of OS and RFS (Tables 1, 2). We also analyzed the association between recurrence pattern and Rubicon expression; 25 patients (89.3%) in the strongly positive group and 29 patients (90.6%) in the weakly positive group had distant metastasis (p = 0.8630). In addition, seven patients (25%) in the strongly positive group and three patients (9.4%) in the weakly positive group had local recurrence (p = 0.1052). Although there was no significant difference, the local recurrence rate was higher in the strongly positive group.

Immunohistochemistry of Rubicon expression. a The cytoplasm and membrane of the splenic pulp are positive controls. b Representative tissue section in human pancreatic ductal adenocarcinoma (PDAC) shows a weaker staining pattern than that observed in the spleen (weakly positive). c Representative tissue section in human PDAC shows a stronger staining pattern than that observed in the spleen endothelial cells (strongly positive). d, e Kaplan–Meier analysis of PDAC patients after neoadjuvant chemoradiotherapy, grouped according to Rubicon expression levels. The strong Rubicon expression group shows significantly lower d overall survival and e recurrence-free survival than the weak Rubicon expression group. Scale bar = 100 µm
MIA-GR cells had higher expression of Rubicon than MIAPaCa-2 cells
The in vitro experiments were performed with MIA-GR cell lines, which were established by long-term exposure to GEM, to validate the immunohistochemistry results, indicating that Rubicon expression affects the prognosis of PDAC after neoadjuvant therapy. We compared the expression level of Rubicon in MIAPaCa-2 and MIA-GR cells with qRT-PCR and WB. MIA-GR cells had higher mRNA and protein expression of Rubicon than MIAPaCa-2 cells (Fig. 2a, b). Moreover, to investigate the mechanism by which MIA-GR cells have higher Rubicon expression, the effect of long-term exposure to GEM on Rubicon in the establishment of the MIA-GR cell line was evaluated. Growth inhibition assay of parent MIAPaCa-2 cells treated with GEM showed that IC70 was 5 ng/mL (Supplementary Fig. 1). MIAPaCa-2 cell line was exposed to 5 ng/mL GEM for about 8 weeks, and qRT-PCR and WB were performed. In both WB and qRT-PCR results using this sample, Rubicon was upregulated in parent MIAPaCa-2 cells (Fig. 2c, d).

Rubicon expression examined by western blotting (WB) and quantitative real-time polymerase chain reaction (qRT-PCR) using MiIAPaCa-2 and MIA-GR, along with the assessment of change in Rubicon expression caused by long-term exposure to gemcitabine (GEM). a qRT-PCR results and b WB analysis show Rubicon mRNA expression in three MIA-GR cell lines (MIA-GR1, MIA-GR2, and MIA-GR3) compared with the expression in MIAPaCa-2. Rubicon expression in MIAPaCa-2 after long-term exposure to GEM shown using c qRT-PCR and d WB. *p < 0.05, **p < 0.01
MIA-GR cells had lower autophagy flux than MIAPaCa-2 cells
In addition to Rubicon, LC3-II and p62 (known as autophagy markers) levels were evaluated using WB. The amount of LC3-II correlates with the number of autophagosomes, and p62 is specifically degraded by autophagy. To evaluate autophagic flux, it is important to measure the amount of LC3-II delivered to lysosomes by comparing LC3-II levels in the presence and absence of lysosomal protease inhibitors (autophagic flux assay). Thus, autophagic flux assay was performed with bafilomycin, a chemical autophagy inhibitor, and the autophagic flux index from the ratio of LC3-II to beta-actin was calculated. The results showed that MIA-GR cells had a lower autophagic flux than parent MIAPaCa-2 cells (p < 0.01) (Fig. 3a, b).

Autophagic flux assay in MIA-GR and MIAPaCa-2 performed using bafilomycin. a Autophagic flux assay in MIA-GR and MIAPaCa-2, b autophagic flux index (the ratio of LC3B-II to beta-actin). **p < 0.01
MIA-GR cells transfected with siRubicon showed no significant change in sensitivity to gemcitabine but exhibited decreased invasion ability and cell viability
Transfection of siRubicon into MIA-GR cells having a higher expression of Rubicon than the parent MIAPaCa-2 cells was performed, confirming that Rubicon expression was reduced in MIA-GR cells transfected with siRubicon using WB (Fig. 4a). In addition, the autophagy flux of MIA-GR cells transfected with siRubicon was evaluated. The results showed that autophagy flux was upregulated when Rubicon expression was suppressed (siScramble vs. siRubicon-1, p = 0.0333; siScramble vs. siRubicon-2, p = 0.0143) (Fig. 4b). Growth inhibitor assay was performed to evaluate the contribution of Rubicon in the mechanism of GEM resistance, but no significant change in GEM sensitivity was shown even with siRubicon knockdown (Fig. 5a). Cell viability and invasion assays were also performed to evaluate the invasion and cell viabilities of MIA-GR cells transfected with siRubicon. The results showed that siRubicon knockdown significantly decreased the cell viability (siScramble vs siRubicon-1, p < 0.01; siScramble vs siRubicon-2, p < 0.01) and invasion ability (siScramble vs siRubicon-1, p = 0.013; siScramble vs siRubicon-2, p < 0.01) of MIA-GR cells (Fig. 5b, c).

Rubicon knockdown in MIA-GR and increase in autophagy flux by knockdown of Rubicon. a Knockdown of Rubicon expression in MIA-GR analyzed using western blotting, b autophagic flux assay in MIA-GR transfected with siRubicon. *p < 0.05

Effect of Rubicon knockdown on gemcitabine (GEM) sensitivity, cell viability, and invasion ability in MIA-GR. a Growth inhibition assay for GEM sensitivity, b cell viability assay, and c invasion assay. Scale bar = 100 µm. *p < 0.05, **p < 0.01
Discussion
It was observed that high expression of Rubicon in patients with PDAC after neoadjuvant chemoradiotherapy could be a prognostic factor. Moreover, in the GR PDAC cell line that survived long-term GEM exposure, Rubicon expression was higher compared to that in the parental cell line. Knockdown of Rubicon expression reduced invasion ability and cell viability.
Autophagy is a lysosomal degradative mechanism characterized by the formation of double-membrane autophagic vesicles called autophagosomes, which engulf portions of cytosol, damaged organelles, and protein aggregates [32]. Considerable studies have already been conducted on the mechanisms of autophagy in cancer and, in recent years, several reports have focused on autophagy in the treatment of pancreatic cancer. However, the role of autophagy in the treatment of pancreatic cancer remains controversial [13, 14, 33]. Although therapeutic development in pancreatic cancer focusing on the mechanism of autophagy is ongoing, it has not been proven sufficiently effective [12].
Rubicon negatively regulates autophagy by interacting with the p13K complex that is essential for autophagy [15, 34]. Although autophagy-regulating proteins, such as the Atg family, promote autophagy, Rubicon is a rare protein that suppresses this process. Rubicon has been associated with NAFLD and cholestasis, along with aging, adipocytes, and Sertoli cells [16,17,18, 35, 36]. In recent years, the association with Rubicon has been reported in cancers of several organs. In human liver cancer cells, Acanthopanax senticosus Harmas extract causes G0/G1 cell cycle arrest and induces autophagy by decreasing Rubicon expression [37]. In addition, combination therapy with mTOR inhibitors and Honokiol induces autophagy via modulation by Rubicon and inhibits the growth of renal tumors after organ transplantation [38]. However, there are no reports on the association of Rubicon with autophagy in pancreatic cancer.
As part of our preliminary research, we investigated Rubicon expression in PDAC cells without neoadjuvant chemoradiotherapy using immunohistochemistry. The 37 PDAC specimens were divided into a strongly positive group (n = 19) and a weakly positive group (n = 18). The Kaplan–Meier analysis showed no significant differences in OS or RFS between the two groups (Log-rank test; OS: p = 0.8435; RFS: p = 0.5796) (Supplementary Fig. 2). Furthermore, there were not any significant differences in the Cox regression analysis either (OS: hazard ratio, 0.99; 95% CI 0.49–2.02; p = 0.9887: RFS: hazard ratio, 1.30; 95% CI 0.62–2.71; p = 0.4870). We have already treated PDAC with neoadjuvant chemoradiotherapy and reported its benefits [39, 40]. Therefore, in this study, we evaluated Rubicon expression in PDAC cells after neoadjuvant chemoradiotherapy. Immunohistochemistry of PDAC cells after neoadjuvant chemoradiotherapy showed that OS and RFS were significantly shorter in the group with high Rubicon expression than in the group with low Rubicon expression. These results suggest that Rubicon may be a prognostic marker in patients with PDAC after neoadjuvant chemoradiotherapy.
Next, we performed in vitro cell experiments with GR PDAC cell lines, which were established by long-term exposure to GEM, as in pancreatic cancer after neoadjuvant therapy. Their specific characteristics in GR PDAC cell lines have been previously reported [41,42,43]. Here, the GR PDAC cell line had higher expression of Rubicon than the parental PDAC cell line. Furthermore, since the difference between the parent and GR cell lines was due to the presence or absence of long-term exposure to GEM, the correlation between long-term exposure to GEM and the expression of Rubicon was investigated. qRT-PCR and WB were performed on samples of the parental cell line exposed to 5 ng/mL GEM for about 8 weeks. The results suggest that long-term exposure to GEM causes upregulation of Rubicon expression in PDAC cells.
Moreover, autophagy flux in GR PDAC and parental cell lines was evaluated. The result showed that the GR cell line had lower autophagy flux than the parental cell line. Since the parental cell lines exhibit relatively high autophagy flux, suppressing the parental cells with an autophagy inhibitor such as an antimalarial drug may have a therapeutic effect. However, because GR cell lines have higher Rubicon expression and lower autophagy flux than parental cell lines, the therapeutic effect of an autophagy inhibitor on GR cell lines may not be as effective as in the parental cell lines. This may be one of the reasons why the abovementioned clinical trials of the HCQ in combination with GEM and nab-PTX of neoadjuvant therapy for PDAC did not show improved prognosis [12].
Since Rubicon was highly expressed in GR cell lines, siRubicon was transfected into GR cell lines and evaluated. Growth inhibition assays were performed to investigate Rubicon in the underlying mechanism of GEM resistance, but there was no significant change in GEM sensitivity in GR cell lines transfected with siRubicon. This result suggests that Rubicon does not contribute to the mechanism of GEM resistance in PDAC.
Malignancy features such as invasion ability and cell viability in siRubicon-transfected GR cell lines were observed to be diminished in this study. Furthermore, autophagy flux was upregulated by siRubicon transfection into the GR cell line. This suggests that autophagy may contribute to the suppression of tumor growth in GR PDAC cell lines. In the past, it has been reported that ubiquitination of UVRAG, which forms a complex with Rubicon, promotes autophagy by autophagosome maturation and inhibits tumor growth in hepatocellular carcinoma [44]. Moreover, the acetylation of Beclin-1 promotes autophagy that degrades tumor growth factors and acts as a tumor suppressor [45]. In PDAC cells that survived neoadjuvant therapy and acquired treatment resistance, Rubicon is elevated and autophagy is suppressed, which may contribute to tumor growth by inhibiting the degradation of tumor growth factors. In addition, immunohistochemistry results showed that the local recurrence rate tended to be higher in the strongly Rubicon-positive group, and the in vitro results of this study showed that Rubicon knockdown in MIA-GR (higher expression of Rubicon than parent cell) reduced invasive ability (Fig. 5c), suggesting a possible involvement of Rubicon in invasive ability.
The elevated expression of Rubicon as a poor prognostic factor in PDAC with NACRT was demonstrated by the results of our in vitro experiments that Rubicon levels were higher in GR PDAC cell lines and were elevated with long-term exposure to GEM. Some PDAC patients without NACRT had high Rubicon expression despite not receiving long-term exposure to GEM. Moreover, high Rubicon expression was not a poor prognostic factor in PDAC patients without NACRT (Supplementary Fig. 2). There may be different functions and mechanisms for each Rubicon between PDAC cases without NACRT with high Rubicon expression and PDAC cases with up-regulated Rubicon after NACRT. The mechanisms and regulators of Rubicon expression have not yet been elucidated, and further studies are needed in this regard.
In conclusion, this study showed that Rubicon may be a prognostic factor and risk factor for PDAC recurrence after neoadjuvant therapy. Rubicon expression was higher in the GR PDAC cell line, which survived long-term GEM exposure than that in the parental cell line, and Rubicon knockdown reduced invasion ability and cell viability. These findings may contribute to the development of novel therapies targeting Rubicon.
Data availability
The data that support the fndings of this study are available on request from the corresponding author, Shogo Kobayashi.
References
Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69:7–34
Muller PC, Frey MC, Ruzza CM et al (2021) Neoadjuvant chemotherapy in pancreatic cancer: an appraisal of the current high-level evidence. Pharmacology 106:143–153
Fujii T, Yamada S, Murotani K et al (2015) Inverse probability of treatment weighting analysis of upfront surgery versus neoadjuvant chemoradiotherapy followed by surgery for pancreatic adenocarcinoma with arterial abutment. Medicine (Baltimore) 94:e1647
Jang JY, Han Y, Lee H et al (2018) Oncological benefits of neoadjuvant chemoradiation with gemcitabine versus upfront surgery in patients with borderline resectable pancreatic cancer: a prospective, randomized, open-label, multicenter phase 2/3 trial. Ann Surg 268:215–222
Eguchi H, Yamada D, Iwagami Y et al (2018) Prolonged neoadjuvant therapy for locally advanced pancreatic cancer. Dig Surg 35:70–76
Colvin H, Mizushima T, Eguchi H et al (2017) Gastroenterological surgery in Japan: the past, the present and the future. Ann Gastroenterol Surg 1:5–10
Rawla P, Sunkara T, Gaduputi V (2019) Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol 10:10–27
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Gotoh K, Lu Z, Morita M et al (2009) Participation of autophagy in the initiation of graft dysfunction after rat liver transplantation. Autophagy 5:351–360
Onorati AV, Dyczynski M, Ojha R et al (2018) Targeting autophagy in cancer. Cancer 124:3307–3318
Hashimoto D, Blauer M, Hirota M et al (2014) Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur J Cancer 50:1382–1390
Zeh HJ, Bahary N, Boone BA et al (2020) A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin Cancer Res 26:3126–3134
Mukubou H, Tsujimura T, Sasaki R et al (2010) The role of autophagy in the treatment of pancreatic cancer with gemcitabine and ionizing radiation. Int J Oncol 37:821–828
Pardo R, Lo Ré A, Archange C et al (2010) Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 10:19–26
Matsunaga K, Saitoh T, Tabata K et al (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396
Tanaka S, Hikita H, Tatsumi T et al (2016) Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 64:1994–2014
Panzitt K, Jungwirth E, Krones E et al (2020) FXR-dependent Rubicon induction impairs autophagy in models of human cholestasis. J Hepatol 72:1122–1131
Nakamura S, Oba M, Suzuki M et al (2019) Suppression of autophagic activity by Rubicon is a signature of aging. Nat Commun 10:847
Goan YG, Zhou B, Hu E et al (1999) Overexpression of ribonucleotide reductase as a mechanism of resistance to 2,2-difluorodeoxycytidine in the human KB cancer cell line. Cancer Res 59:4204–4207
Davidson JD, Ma L, Flagella M et al (2004) An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res 64:3761–3766
Nakahira S, Nakamori S, Tsujie M et al (2007) Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer 120:1355–1363
Yamada D, Kobayashi S, Yamamoto H et al (2012) Role of the hypoxia-related gene, JMJD1A, in hepatocellular carcinoma: clinical impact on recurrence after hepatic resection. Ann Surg Oncol 19:S355–S364
Kondo M, Yamamoto H, Nagano H et al (1999) Increased expression of COX-2 in nontumor liver tissue is associated with shorter disease-free survival in patients with hepatocellular carcinoma. Clin Cancer Res 5:4005–4012
Yamamoto H, Kondo M, Nakamori S et al (2003) JTE-522, a cyclooxygenase-2 inhibitor, is an effective chemopreventive agent against rat experimental liver fibrosis. Gastroenterology 125:556–571
Mitsufuji S, Iwagami Y, Kobayashi S et al (2022) Inhibition of clusterin represses proliferation by inducing cellular senescence in pancreatic cancer. Ann Surg Oncol 29:4937–4946
Mizushima N, Yoshimori T (2007) How to interpret LC3 immunoblotting. Autophagy 3:542–545
Klionsky DJ, Abdelmohsen K, Abe A et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12:1–222
Shiode Y, Hikita H, Tanaka S et al (2020) Hepatitis C virus enhances Rubicon expression, leading to autophagy inhibition and intracellular innate immune activation. Sci Rep 10:15290
Eguchi H, Nagano H, Yamamoto H et al (2000) Augmentation of antitumor activity of 5-fluorouracil by interferon alpha is associated with up-regulation of p27Kip1 in human hepatocellular carcinoma cells. Clin Cancer Res 6:2881–2890
Mukai Y, Yamada D, Eguchi H et al (2018) Vitamin D supplementation is a promising therapy for pancreatic ductal adenocarcinoma in conjunction with current chemoradiation therapy. Ann Surg Oncol 25:1868–1879
Shinke G, Yamada D, Eguchi H et al (2018) Role of histone deacetylase 1 in distant metastasis of pancreatic ductal cancer. Cancer Sci 109:2520–2531
Amaravadi RK, Lippincott-Schwartz J, Yin XM et al (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17:654–666
Yang S, Wang X, Contino G et al (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev 25:717–729
Zhong Y, Wang QJ, Li X et al (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 11:468–476
Yamamuro T, Kawabata T, Fukuhara A et al (2020) Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat Commun 11:4150
Yamamuro T, Nakamura S, Yamano Y et al (2021) Rubicon prevents autophagic degradation of GATA4 to promote Sertoli cell function. PLoS Genet 17:e1009688
Kawano Y, Tanaka M, Fujishima M et al (2021) Acanthopanax senticosus harms extract causes G0/G1 cell cycle arrest and autophagy via inhibition of Rubicon in human liver cancer cells. Oncol Rep 45:1193–1201
Sabarwal A, Wedel J, Liu K et al (2021) A combination therapy using mTOR inhibitor and Honokiol effectively induces autophagy through the modulation of AXL and Rubicon in renal cancer cells, and restricts renal tumor growth following an organ transplantation. Carcinogenesis 43:360–370
Eguchi H, Takeda Y, Takahashi H et al (2019) A prospective, open-label, multicenter phase 2 trial of neoadjuvant therapy using full-dose gemcitabine and S-1 concurrent with radiation for resectable pancreatic ductal adenocarcinoma. Ann Surg Oncol 26:4498–4505
Takeda Y, Nakamori S, Eguchi H et al (2014) Neoadjuvant gemcitabine-based accelerated hyperfractionation chemoradiotherapy for patients with borderline resectable pancreatic adenocarcinoma. Jpn J Clin Oncol 44:1172–1180
Kubo M, Gotoh K, Eguchi H et al (2020) Impact of CD36 on chemoresistance in pancreatic ductal adenocarcinoma. Ann Surg Oncol 27:610–619
Mikamori M, Yamada D, Eguchi H et al (2017) MicroRNA-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci Rep 7:42339
Iwagami Y, Eguchi H, Nagano H et al (2013) miR-320c regulates gemcitabine-resistance in pancreatic cancer via SMARCC1. Br J Cancer 109:502–511
Feng X, Jia Y, Zhang Y et al (2019) Ubiquitination of UVRAG by SMURF1 promotes autophagosome maturation and inhibits hepatocellular carcinoma growth. Autophagy 15:1130–1149
Sun T, Li X, Zhang P et al (2015) Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun 6:7215
Acknowledgements
The authors would like to thank Editage (www.editage.com) for English language editing.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
DM, KG, SK, and HE designed the study. DM, KG, SK, KS, YI, DY, YT, HA, TA, TN, HT, MT, YD, and HE drafted the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10147_2023_2306_MOESM1_ESM.tif
Supplementary file1 Growth inhibition assay of parent MIAPaCa-2 cells treated with gemcitabine with the IC70 value at 5 ng/mL (TIF 1378 KB)
10147_2023_2306_MOESM2_ESM.tif
Supplementary file2 Kaplan–Meier analysis of PDAC patients without neoadjuvant chemoradiotherapy, grouped according to Rubicon expression levels. There were no significant differences in OS or RFS between the two groups (TIF 1473 KB)
About this article

Cite this article
Marukawa, D., Gotoh, K., Kobayashi, S. et al. Rubicon can predict prognosis in patients with pancreatic ductal adenocarcinoma after neoadjuvant chemoradiotherapy. Int J Clin Oncol 28, 576–586 (2023). https://doi.org/10.1007/s10147-023-02306-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-023-02306-0