
Abstract
Background
The randomized, double-blind, placebo-controlled GRID trial tested the oral multikinase inhibitor regorafenib in 199 patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib, and showed a significant improvement in progression-free survival (PFS) versus placebo [hazard ratio (HR) 0.27; 95 % confidence interval (CI) 0.19–0.39; p < 0.0001].
Methods
A subgroup analysis of Japanese patients in the GRID study was performed to compare the efficacy and safety of oral regorafenib 160 mg once daily with matching placebo, in combination with best supportive care. The primary study endpoint was progression-free survival (PFS); safety was evaluated through the incidence of adverse events (AEs).
Results
Seventeen Japanese patients were randomized to regorafenib (n = 12) or placebo (n = 5). Patient demographics were consistent with those of the overall study population. PFS was significantly longer with regorafenib than placebo (HR 0.08; 95 % CI 0.02–0.45; p = 0.000164). Centrally assessed disease control rates were 58 % and 20 % in the regorafenib and placebo groups, respectively (p = 0.080796). Treatment-related adverse events (AEs) were reported in all regorafenib-treated patients and 60 % of placebo recipients; the most frequent AE was hand–foot skin reaction (HFSR) (92 % versus 20 %, respectively).
Conclusion
Regorafenib showed efficacy and a manageable safety profile in Japanese patients with advanced GIST, consistent with the overall GRID study population. AEs, such as HFSR and maculopapular rash, were observed more frequently in Japanese patients. Although dose modification was frequently reported, only one patient with hepatic failure discontinued regorafenib because of AEs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumors (GIST) are the most commonly occurring sarcomas [1]. Although the incidence of GIST in Japan has not been reported, studies conducted in several countries show an estimated rate of 10–20 new cases per year per million population [2–4]. As the incidence of GIST is not thought to differ between ethnicities [4], these data suggest an incidence in Japan of approximately 1,500–2,500 cases per year.
GIST are thought to originate from interstitial cells of Cajal, and one of their defining features is the presence of oncogenic gain-of-function mutations in the genes encoding KIT and platelet-derived growth factor receptor (PDGFR) A [1]. KIT mutations are present in approximately 75–80 % of GIST, whereas PDGFRα mutations are present in 5–8 % of tumors [5], and these mutations are thought to drive the growth of GIST. The accepted first- and second-line therapies for GIST are imatinib and sunitinib, respectively, both of which inhibit KIT and PDGFRα [6, 7]. In addition, sunitinib inhibits angiogenesis by acting on vascular endothelial growth factor receptors (VEGFRs) [6, 7]. Although imatinib and sunitinib have demonstrated efficacy in a number of clinical studies [7], most tumors develop resistance to treatment as a result of secondary mutations in KIT and PDGFRα [7, 8] and, until recently, no third-line therapy was available for patients with GIST refractory to imatinib and sunitinib therapy.
Regorafenib is an orally administered multikinase inhibitor with activity against protein kinases associated with angiogenesis (VEGFR 1–3 and TIE2), oncogenesis (KIT, RET, RAF-1, and wild-type and V600E-mutated BRAF), and maintenance of the tumor microenvironment (PDGFR and fibroblast growth factor receptor) [9]. The efficacy of regorafenib has been demonstrated in the phase III GRID trial involving patients with advanced GIST in which imatinib and sunitinib had failed [10]. On the basis of these results, regorafenib was approved in this indication by the U.S. Food and Drug Administration in February 2013 [11] and by the Japanese Ministry of Health, Labor, and Welfare in August 2013 [12].
In the GRID study, patients were randomized to receive either regorafenib (n = 133) or placebo (n = 66) in addition to best supportive care (BSC) [10]. The trial met its primary endpoint of centrally assessed progression-free survival (PFS), with a hazard ratio (HR) for regorafenib versus placebo of 0.27 [95 % confidence interval (CI) 0.19–0.39; p < 0.0001]. Median PFS was 4.8 months (95 % CI 4.0–5.7) in the regorafenib group and 0.9 (95 % CI 0.9–1.05) in the placebo group. Overall survival (OS) did not differ significantly between groups, with a HR for regorafenib versus placebo of 0.77 (95 % CI 0.42–1.41; p = 0.199). Adverse events (AEs) were reported in all regorafenib-treated patients and in 92 % of placebo recipients during double-blind treatment, and treatment-related AEs were reported in 99 % and 68 %, respectively. The most common treatment-related AEs observed in regorafenib-treated patients were hand–foot skin reaction (HFSR), hypertension, diarrhea, fatigue, and oral mucositis.
There is increasing evidence that kinase inhibitors have differing tolerability profiles in Asian versus non-Asian patients. In studies assessing sunitinib in patients with advanced renal cell carcinoma (RCC), Korean and Japanese investigators independently reported that patients in their countries showed clinical efficacy that was at least as good as that seen in global studies, although incidences of hematological AEs were higher than those observed in Western patients [13, 14]. Similarly, analysis of a study comparing axitinib and sorafenib in patients with metastatic RCC showed a higher incidence of some AEs in Japanese patients than in the overall study population [15]. Although analysis of the overall GRID study population showed no significant variation in the efficacy of regorafenib across geographic regions, variations in the tolerability profile of regorafenib between populations have not been assessed [10]. In light of the approval of regorafenib in Japan, we performed a post hoc subgroup analysis to assess the efficacy and safety of regorafenib in Japanese patients enrolled in the GRID study.
Patients and methods
GRID was a randomized, double-blind, multicenter, parallel-group trial involving 199 patients from 57 centers in 17 countries in Asia, Europe, and North America, including 17 Japanese patients. Details of patient inclusion and exclusion criteria have been published previously [10]. Briefly, eligible patients were at least 18 years of age, with histologically confirmed metastatic or unresectable GIST following failure of previous treatment with at least imatinib (caused by either disease progression or intolerance) and sunitinib (caused solely by progression, because the definition of intolerance is more variable with this agent). Patients were also allowed to have received other approved and investigational systemic therapy for GIST. Other inclusion criteria of note were at least one measurable lesion on computed tomography or magnetic resonance imaging according to modified Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1; resolution of all toxicities from previous therapies to at least National Cancer Institute Common Terminology Criteria for Adverse Events grade 1; Eastern Cooperative Oncology Group performance status of 0 or 1; and adequate hematological, hepatic, cardiac, and renal function. Criteria for excluding patients from the trial included previous treatment with a VEGFR inhibitor other than sunitinib and uncontrolled hypertension (systolic blood pressure >140 mmHg or diastolic blood pressure >90 mmHg despite optimal medical treatment).
The study protocol was approved by each participating center’s institutional review board. The trial followed the principles of the Declaration of Helsinki and good clinical practice and complied with all local laws. All patients provided written informed consent before enrollment. The trial was registered with ClinicalTrials.gov, identifier NCT01271712.
Treatment
Patients were randomized to receive oral regorafenib 160 mg or matching placebo once daily for weeks 1–3 of each 4-week cycle, in combination with BSC. Randomization was stratified by treatment line (failure of imatinib and sunitinib versus failure of imatinib, sunitinib, and other GIST therapies) and geographic region (Asia versus rest of world). BSC included antibiotics, analgesics, bisphosphonates, corticosteroids, transfusions, or other symptomatic therapy, but excluded other investigational agents, antineoplastic drugs, surgical intervention, palliative radiotherapy, hormonal therapy, and biological response modifiers such as granulocyte colony-stimulating factor. Protocol-specified dose interruptions and reductions were allowed for the management of clinically relevant, treatment-related toxicities; details of the dose modification protocols have been published previously [10]. Regorafenib dosage could be re-escalated to a maximum of 160 mg/day at the investigator’s discretion after toxicities had resolved to prespecified levels. Treatment was permanently discontinued in patients whose toxicities failed to resolve after a 4-week dose interruption or a reduction of two consecutive dose levels (to 80 mg/day).
Patients continued blinded treatment until disease progression, death, unacceptable toxicity, or withdrawal from the study. Tumors were assessed every 4 weeks for the first 3 months of the trial, every 6 weeks from months 3 to 6, and then every 8 weeks until the end of treatment. Treatment was unblinded at the time of centrally assessed disease progression, at which point patients in either group were allowed to receive optional open-label regorafenib treatment, depending on physician discretion.
Endpoints
The primary endpoint of the trial was PFS, assessed by two central radiology reviewers using modified RECIST version 1.1 [10]. Disagreements between the two reviewers were adjudicated by a third radiology reviewer. Secondary endpoints included OS, time to disease progression, objective response rate, and disease control rate (DCR; defined as complete response, partial response, or stable disease of ≥12 weeks duration), which were also assessed centrally. A secondary investigator assessment of PFS was also performed in patients who received regorafenib in the open-label follow-up period after centrally assessed disease progression during double-blind treatment with regorafenib or placebo (secondary PFS). Safety was assessed by review of AEs and laboratory data.
Statistical analysis
The GRID trial was designed to have 94 % power to detect a 100 % improvement in PFS in the regorafenib group versus the placebo group, based on a patient population of 199 patients randomized to receive regorafenib and placebo in a 2:1 ratio, with a total of 144 events needed for the final PFS analysis. Statistical analyses were performed with SAS version 9.1 or higher (SAS, Cary, NC, USA).
Efficacy analyses were performed using the intention-to-treat population; the safety population included all patients who received at least one dose of study drug. Post hoc analyses were performed in the Japanese subgroup and were compared with outcomes in the overall GRID study population. PFS and OS estimates were calculated using the Kaplan–Meier method, with HRs and 95 % CIs derived from a Cox proportional hazards model, and p values obtained with a stratified log-rank test. Objective response rates and DCRs were compared between groups with the Cochran–Mantel–Haenszel test.
Results
Twenty-one patients were screened for inclusion in the GRID trial at six centers in Japan; 17 patients met the patient selection criteria and were randomized to receive regorafenib (n = 12) or placebo (n = 5), with all patients receiving at least one dose of study drug. Six patients in the regorafenib group and four patients in the placebo group received open-label regorafenib following disease progression. The flow of Japanese patients through the GRID trial is shown in Fig. 1. The baseline characteristics and demographics of the Japanese subgroup and the overall GRID patient population are shown in Table 1.

Flow of Japanese patients through the GRID study
The mean ± standard deviation duration of treatment in the double-blind period was 23.1 ± 13.4 weeks (median 22.9; range 5.7–42.9) in the regorafenib group and 7.9 ± 6.0 weeks (median 7.0; range 3.0–18.2) in the placebo group. Mean ± standard deviation daily dosage of study drug in the double-blind period was 126.1 ± 20.4 mg (median 124.0; range 95.3–160.0) in the regorafenib group and 160.0 ± 0.0 mg (median 160.0) in the placebo group. Dose modifications were reported in 1 placebo recipient (20 %) and 11 (92 %) regorafenib-treated patients during double-blind treatment, consisting of dose reductions in 2 regorafenib-treated patients (17 %) and dose interruptions in 11 regorafenib-treated patients (92 %) and the 1 placebo recipient (20 %).
Efficacy
Median centrally assessed PFS was longer in the regorafenib group than in the placebo group in Japanese patients, with a HR versus placebo of 0.08 (95 % CI 0.02–0.45; one-sided p = 0.000164; Fig. 2). Median durations of PFS in the Japanese subgroup were 0.9 months (95 % CI 0.69–2.70) in placebo recipients and 7.1 months (95 % CI 2.76 to not estimable) in regorafenib-treated patients. Median investigator-assessed PFS in the placebo group was 0.9 months (95 % CI 0.7 to not estimable); the median investigator-assessed PFS for the regorafenib group could not be estimated, with a lower limit of the 95 % CI of 7.1 months. OS did not differ significantly between groups (HR versus placebo of 0.42; 95 % CI 0.06–2.95; one-sided p = 0.182; Fig. 3). Secondary PFS during open-label treatment did not differ between treatment groups, with median durations of 4.5 months (95 % CI 2.53–4.93) and 5.4 months (95 % CI 1.38–5.36) in patients originally randomized to regorafenib and placebo, respectively.

Kaplan–Meier plot of progression-free survival in Japanese patients. CI confidence interval, HR hazard ratio

Kaplan–Meier plot of overall survival in Japanese patients. CI confidence interval, HR hazard ratio
In Japanese patients, central assessment showed no complete response in either treatment group, although partial response was observed in 1 patient (20 %) in the placebo group. Stable disease was observed in 11 patients (92 %) in the regorafenib group, and no patients in the placebo group, with disease control observed in 7 patients (58 %) and 1 patient (20 %), respectively (difference from placebo 38 %; 95 % CI 11–87; p = 0.080796).
Centrally assessed reductions of target lesion volumes were observed in six patients (50 %) in the regorafenib group and one patient (20 %) in the placebo group, with three patients (25 %) in the regorafenib group and one patient (20 %) in the placebo group having a reduction in target lesion volume of at least 10 % (Fig. 4). Tumor growth was observed in five patients (42 %) in the regorafenib group and four patients (80 %) in the placebo group.

Maximum change in target lesion size in Japanese patients
Safety and tolerability
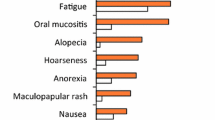
In the Japanese subgroup, treatment-emergent AEs of any grade were reported in all patients receiving regorafenib or placebo, with serious AEs reported in three regorafenib-treated patients (25 %) and one placebo recipient (20 %). AEs considered to be treatment related were observed in all regorafenib-treated patients and in three patients (60 %) in the placebo group (Table 2). Grade 3 or greater treatment-related AEs were observed in ten regorafenib-treated patients (83 %) and no placebo recipients. The most frequently observed grade 3 or greater treatment-related AEs in the regorafenib group were hypertension (three patients, 25 %), HFSR (two patients, 17 %), maculopapular rash (two patients, 17 %), and decreased neutrophil count (two patients, 17 %). The most frequent AEs requiring dose interruption in regorafenib-treated patients were HFSR (seven patients, 58 %), maculopapular rash (two patients, 17 %), and fatigue (two patients, 17 %). The only AE requiring regorafenib dose reduction in more than one patient was HFSR, reported in five patients (42 %). One patient, a 49-year-old man with metastatic GIST, discontinued regorafenib treatment as a result of acute hepatic failure that was considered to be drug related. The patient was hospitalized 9 days after beginning his second cycle of treatment (6 weeks after the start of treatment) with symptoms of hepatic failure, and regorafenib was discontinued; the patient died 2 weeks later.
Discussion
The international GRID study showed a significant improvement in centrally assessed PFS in regorafenib-treated patients versus placebo recipients with advanced GIST after failure of imatinib and sunitinib [10]. Following disease progression, 85 % of placebo recipients crossed over to receive regorafenib, resulting in no significant difference in OS between groups. A best response of partial response or stable disease was observed in 76 % of regorafenib-treated patients and 35 % of placebo recipients.
The Japanese subgroup had similar baseline characteristics to those of the overall study population. Outcomes in the Japanese patients were generally consistent with those observed in the overall GRID trial. Although median PFS in the Japanese placebo population was the same as that observed in the overall placebo population (0.9 months), regorafenib-treated patients in both the Japanese and overall study populations had prolonged PFS versus placebo recipients (median PFS, 7.1 and 4.8 months, respectively). As with the overall population, the difference in OS between regorafenib and placebo recipients was not significant in the Japanese population, likely as a result of crossover. Centrally assessed partial response was infrequent in both the Japanese subgroup and the overall GRID population, whereas stable disease rates appeared higher in the Japanese subgroup (92 %) than in the overall trial population (71 %). DCRs in regorafenib-treated patients were similar in the Japanese subgroup (58 %) and the overall GRID population (53 %). Secondary PFS during open-label treatment after progression was generally similar between the Japanese regorafenib and placebo subgroups and the overall GRID regorafenib and placebo groups. In addition, secondary PFS in the Japanese subgroups appeared somewhat similar to centrally assessed PFS during double-blind treatment with regorafenib in both the Japanese subgroup and the overall study population. This outcome may be partly because of tumor heterogeneity, which could result in a subset of tumor cells remaining sensitive to regorafenib, despite an overall outcome of disease progression. This finding is consistent with the results of a study conducted by Agaram et al. [16], which showed heterogeneous histological responses to imatinib treatment in GIST cells. In addition, an analysis of genetic biomarkers in patients enrolled in the GRID study showed a higher prevalence of KIT mutations associated with loss of responsiveness to kinase inhibitors in circulating tumor DNA compared with archival tissue, providing further evidence for the presence of tumor heterogeneity in the GRID study population [17].
In the overall GRID population, treatment-related AEs were reported in 99 % of regorafenib-treated patients and 68 % of placebo recipients. The most common grade 3 or higher regorafenib-related AEs were hypertension, HFSR, and diarrhea. The safety profile of regorafenib in Japanese patients was similar to that in the overall study population, although the frequency of some AEs in Japanese patients differed from that in the overall study population. Regorafenib-related HFSR of any grade was reported more frequently in Japanese patients (92 %) than in the overall population (56 %). This finding is consistent with previous trials of kinase inhibitors, which have shown a higher incidence of HFSR in Asian versus non-Asian patients [18]. However, rates of grade 3 HFSR were similar in the Japanese population (17 %) and overall populations (20 %). Maculopapular rash was reported in 50 % of regorafenib-treated Japanese patients, compared with 18 % of regorafenib-treated patients in the overall GRID population, with grade 3 or greater maculopapular rash reported in 17 % and 2 % of patients, respectively. Other AEs associated with regorafenib, such as diarrhea, fatigue, and hypertension, occurred at similar rates in the Japanese and overall study populations.
A numerically higher rate of some regorafenib-related AEs, including HFSR, was also observed in a subgroup analysis of Japanese patients enrolled in the CORRECT trial, which compared the outcomes of Japanese and non-Japanese patients [19]. However, in contrast to the current analysis, no marked elevation in the rate of rash or desquamation was observed, although the rate of diarrhea was lower in Japanese than in non-Japanese patients [19]. In addition, the rates of grade 3 or higher HFSR were similar in Japanese and non-Japanese patients. The difference in adverse event rates led the authors to further investigate the relationship between regorafenib-related AEs and patient characteristics by performing analyses in subgroups based on body surface area (BSA) and body mass index (BMI). However, no clear relationship between the incidence of AEs and baseline BMI or BSA was observed [19].
The safety profile of regorafenib in Asian patients has been further elaborated in the CONCUR trial, which randomized 204 patients with metastatic colorectal cancer in five Asian countries [20]. In a preliminary analysis of AEs, treatment-emergent HFSR of any grade was observed in 74 % of regorafenib-treated patients, a rate similar to that in the current study and the Japanese subgroup analysis of CORRECT. Grade 3 or higher HFSR occurred at a similar rate in the CONCUR and CORRECT trials [20]. Rates of other AEs, including maculopapular rash, were generally similar to those in the overall GRID and CORRECT populations [20].
Regorafenib dose modifications (interruptions or reductions), which were used in a greater proportion of Japanese patients (92 %) than in the overall GRID population (72 %), allowed Japanese patients to continue therapy despite the occurrence of AEs. Although the rates of some AEs were higher in the Japanese subgroup than in the overall population, the frequency of AEs leading to permanent discontinuation was low for both populations (6 % each). The mean treatment duration with regorafenib in the Japanese patients (23 weeks) was similar to that observed in the overall population (20 weeks).
Interpretation of this subgroup analysis is subject to some limitations. First, this was not a prespecified subgroup analysis of the GRID trial and thus could be prone to selection bias. Second, only a small number of patients were enrolled at Japanese centers, and this assessment, therefore, lacks sufficient statistical power to draw definitive conclusions. Nonetheless, the demographics and characteristics of the Japanese population were generally consistent with those of the overall GRID population, and, with the exception of the highlighted differences in the safety profile of regorafenib, the outcomes observed in the Japanese population were similar to those of the overall study population. Importantly, however, as only a small number of Japanese patients were included in the GRID study, clinical experience (including from the ongoing Japanese post-marketing surveillance study, ClinicalTrials.gov identifier NCT01933958) will be vital in providing a clear picture of the safety profile of regorafenib in Japanese patients.
References
Blay JY, Le Cesne A, Cassier PA et al (2012) Gastrointestinal stromal tumors (GIST): a rare entity, a tumor model for personalized therapy, and yet ten different molecular subtypes. Discov Med 13:357–367
Nilsson B, Bumming P, Meis-Kindblom JM et al (2005) Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era: a population-based study in western Sweden. Cancer (Phila) 103:821–829
Tryggvason G, Gislason HG, Magnusson MK et al (2005) Gastrointestinal stromal tumors in Iceland, 1990–2003: the Icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer 117:289–293
Nishida T, Takahashi T, Miyazaki Y (2009) Gastrointestinal stromal tumor: a bridge between bench and bedside. Gastric Cancer 12:175–188
Corless CL, Barnett CM, Heinrich MC (2011) Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer 11:865–878
Demetri GD, Benjamin RS, Blanke CD et al (2007) NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST): update of the NCCN clinical practice guidelines. J Natl Comp Canc Netw 5:S1–S29
Blay JY (2011) A decade of tyrosine kinase inhibitor therapy: historical and current perspectives on targeted therapy for GIST. Cancer Treat Rev 37:373–384
Demetri GD, von Mehren M, Antonescu CR et al (2010) NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Comp Canc Netw 8(suppl 2):S1–S41
Wilhelm SM, Dumas J, Adnane L et al (2011) Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 129:245–255
Demetri GD, Reichardt P, Kang Y-K et al (2013) Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381:295–302
US Food and Drug Administration (2013) FDA news release: FDA approves Stivarga for advanced gastrointestinal stromal tumors. US FDA. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm340958.htm. Accessed Aug 2014
Bayer HealthCare (2013) Bayer’s Stivarga® approved for the treatment of patients with gastrointestinal stromal tumors in Japan. http://press.healthcare.bayer.com/en/press/auth/news-details-page.php/15176/2013-0432. Accessed Aug 2014
Kim HS, Hong MH, Kim K et al (2011) Sunitinib for Asian patients with advanced renal cell carcinoma: a comparable efficacy with different toxicity profiles. Oncology 80:395–405
Uemura H, Shinohara N, Yuasa T et al (2010) A phase II study of sunitinib in Japanese patients with metastatic renal cell carcinoma: insights into the treatment, efficacy and safety. Jpn J Clin Oncol 40:194–202
Ueda T, Uemura H, Tomita Y et al (2013) Efficacy and safety of axitinib versus sorafenib in metastatic renal cell carcinoma: subgroup analysis of Japanese patients from the global randomized phase 3 AXIS trial. Jpn J Clin Oncol 43:616–628
Agaram NP, Besmer P, Wong GC et al (2007) Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res 13:170–181
Demetri GD, Jeffers M, Reichardt P et al (2013) Detection of oncogenic kinase mutations in circulating plasma DNA and correlation with clinical benefit in the phase III GRID study of regorafenib vs. placebo in TKI-refractory metastatic GIST. Cancer Res 73:LB–295 (abstract)
Cheng AL, Kang YK, Chen Z et al (2009) Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10:25–34
Yoshino T, Komatsu Y, Yamada Y et al (2014) Randomized phase III trial of regorafenib in metastatic colorectal cancer: analysis of the CORRECT Japanese and non-Japanese subpopulations. Invest New Drugs (Epub ahead of print, 12 Sep 2014)
Li J, Qin S, Yau T et al (2014) CONCUR: a randomized, double-blind, placebo-controlled phase 3 study of regorafenib monotherapy in Asian patients with previously treated metastatic colorectal cancer (mCRC). Ann Oncol 25(Suppl 2):ii114–ii115
Acknowledgments
We thank the participating patients and staff at each of the study centers. Editorial assistance in the preparation of this manuscript was provided by Succinct Medical Communications, with financial support from Bayer Health Care Pharmaceuticals; the authors retained editorial control over the content.
Conflict of interest
Yoshito Komatsu has received honoraria and research funding from Bayer. Yasuhide Yamada has received honoraria from Taiho, Chugai, and Pfizer and research funding from Novartis, Astrazeneca, Otsuka, MerckSerono, Chugai, and Daiichi-Sankyo. Iris Kuss owns stocks in Bayer. George Demetri has acted as a consultant to Kolltan Pharmaceuticals, Blueprint Medicines, G1 Therapeutics, Caris, Champions Oncology, and Bessor Pharmaceuticals (uncompensated), Pfizer, EMD-Serono, Sanofi Oncology, Janssen Oncology, Glaxo-Smith-Kline, Ariad, Astra-Zeneca, WIRB Copernicus Group, and Bayer. He has received research support from Bayer, Novartis, Pfizer, EMD-Serono, Sanofi Oncology, Janssen Oncology and Glaxo-Smith-Kline. He has equity in Kolltan Pharmaceuticals, Blueprint Medicines, G1 Therapeutics, Caris, Champions Oncology, and Bessor Pharmaceuticals. He has been part of scientific advisory boards for Kolltan Pharmaceuticals, Blueprint Medicines, G1 Therapeutics, Caris, and Champions Oncology and has provided noncompensated expert regulatory testimony on behalf of Bayer and Glaxo-Smith-Kline. Toshirou Nishida has received honoraria from Novartis, Pfizer, and Bayer and a research grant from Novartis. Toshihiko Doi, Akira Sawaki, and Tatsuo Kanda have no relevant conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Komatsu, Y., Doi, T., Sawaki, A. et al. Regorafenib for advanced gastrointestinal stromal tumors following imatinib and sunitinib treatment: a subgroup analysis evaluating Japanese patients in the phase III GRID trial. Int J Clin Oncol 20, 905–912 (2015). https://doi.org/10.1007/s10147-015-0790-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-015-0790-y