
Abstract
To investigate the m6a-related long non-coding RNAs (lncRNAs) that may be exploited as potential biomarkers in primary glioblastoma (pGBM), a cohort of 268 glioma samples from GSE16011 dataset was included for discovery. The Chinese Glioma Genome Atlas (CGGA) microarray and RNA sequencing databases were used for validation. Bioinformatic analyses were performed using the R software. The m6a-lncRNA co-expression networks were constructed, and four m6a-related lncRNAs (MIR9-3HG, LINC00900, MIR155HG, and LINC00515) were identified in pGBM patients on the univariate Cox regression analysis. Patients in the low-risk group had longer overall survival (OS) and progression-free survival (PFS) than those in the high-risk group (P = 0.0025, P = 0.0070). Moreover, the high-risk group displayed older age, isocitrate dehydrogenase (IDH) wild-type, classical and mesenchymal TCGA subtype, and G3 CGGA subtype. Distinct m6a status was identified according to histologic grade and two groups (low-risk and high-risk). Functional annotation showed that differentially expressed genes between the two groups were enriched in immune response, apoptosis, cell adhesion, negative regulation of transcription, negative regulation of RNA metabolic process, and regulation of RNA metabolic process. We profiled the m6a status in glioma and identified four m6a-related prognostic lncRNAs for pGBMs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary glioblastoma (pGBM) is a common malignant brain tumor, and the median overall survival (OS) is approximately 14.4 months [1]. It reported that N6-methyladenosine (m6a) is a chemical modification present in multiple RNA species [2]. Many studies have revealed its essential roles in physiological processes. METTL14 is highly expressed in normal hematopoietic stem cells, and it can promote self-renewal of leukemia stem cells and plays an oncogenic role in the development and maintenance of acute myeloid leukemia (AML) [3]. Moreover, WTAP has also been reported to be upregulated in AML [4]. Inhibition of FTO could suppress the growth and self-renewal of glioblastoma stem cell (GSC) and prolongs the lifespan of glioblastoma stem cell-grafted mice [5]. Knockdown FTO could effectively inhibit cell proliferation and invasion in lung squamous cell carcinoma [6]. Overexpression of ALKBH5 could promote the proliferation and tumorigenesis of GSCs [7]. It was reported that YTHDC1 could recognize m6a on XIST and promote X chromosome silencing [8]. YTHDF1 was correlated with the survival and pathology stage in hepatocellular carcinoma [9]. Dysregulation of long non-coding RNAs (lncRNAs) may contribute to glioma pathogenesis [10,11,12,13]. HOXA11-AS and MALAT1 were involved in cell cycle progression and were closely associated with poor prognosis [14, 15]. It indicated that lncRNA GAS5 expressions are negatively correlated with YTHDF3 in tumors, and YTHDF3 could facilitate m6a-modified lncRNA GAS5 degradation [16]. m6a can regulate the expression of lncRNA RP11, then trigger the dissemination of tumor cells [17]. m6a eraser ALKBH5 can demethylate KCNK15-AS1 and regulate KCNK15-AS1-mediated cell motility [18]. Therefore, aberrant expressions of m6a-related lncRNAs may have prognostic value for pGBM patients.
We used the GSE16011 (training set), Chinese Glioma Genome Atlas (CGGA) microarray, and CGGA RNA sequencing (validated sets) databases to explore the prognostic value of m6a-related lncRNAs.
Methods
Patients and datasets
The GSE16011 was downloaded from the Gene Expression Omnibus (GEO). All CGGA data used in this study were available from CGGA website (http://www.cgga.org.cn). The grade 1 samples were excluded to avoid data bias. A total of 757 patients (GSE16011: LGG 109, pGBM 159; CGGA microarray: LGG 152, pGBM 109; CGGA sequencing: LGG 141, pGBM 87) were included in our study. In addition, patients with clinical information were used in the survival analysis. All patients were diagnosed histologically by two neuropathologists according to 2007 WHO classification guidelines. Informed consent was undertaken from the patients, and our study was approved by the Ethics Committee of Beijing Tiantan Hospital. The clinical information was downloaded from each website.
lncRNA profile mining
We identified the lncRNA profiles by using an established lncRNA mining method [19, 20]. Some lncRNA-specific probes were fortuitously represented on the gene microarrays, so we could achieve the lncRNA profiling by analyzing these existing data. For example, lncRNA expression signatures may be determined by mining the Affymetrix gene expression microarray data [21, 22]. The platform of GSE16011 was the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array. Only lncRNAs were retained based on their Refseq IDs or Ensembl IDs. A total of 1248 lncRNAs (1901 probes) were screened from the GSE16011 dataset. The functions of m6a on RNA are determined by the interaction between its methyltransferases, demethylases, and m6a readers [2]. Finally, 493 m6a-related lncRNAs (694 probes) were identified by constructing m6a-lncRNA co-expression networks (Cor value 0.4; P ≤ 0.001).
Statistical analysis
The risk score was determined as previously reported [23,24,25]. Briefly, the risk score was assigned according to a linear combination of the expression level of lncRNAs weighted by the regression coefficient [20, 26, 27].
The prognostic m6a-related lncRNAs were identified using univariate Cox regression analysis and ranked ascendingly by P values. Low-risk and high-risk groups were divided using the median risk score. The multivariate Cox regression analysis was performed by the SPSS software (version 22; SPSS Inc., Chicago, IL, USA), and the principal component analysis (PCA) was performed by the R software (version 3.2.3). GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA) was used to display the Kaplan-Meier curve. DAVID database (https://david.ncifcrf.gov/) was used for functional annotation between two groups. A two-sided P value of < 0.05 was considered statistically significant. The P value has been corrected by false discovery rate (FDR).
Results
The m6a-related genes were dysregulated in gliomas
A total of 16 m6a-related genes were identified from the three datasets, and we analyzed these data to explore the expression patterns of m6a genes in gliomas. As shown in Fig. 1, fourteen m6a-related genes (METTL3, METTL14, WTAP, KIAA1429, RBM15, RBM15B, ZC3H13, FTO, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1, and HNRNPC) were differentially expressed in gliomas comparing with normal brain tissues. Of these genes, METTL3, METTL14, WTAP, RBM15, RBM15B, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPA2B1, and HNRNPC were upregulated, while ZC3H13 and FTO were downregulated in gliomas (Fig. 1). WTAP, RBM15, ALKBH5, and YTHDF2 were significantly upregulated, while METTL3, KIAA1429, ZC3H13, FTO, YTHDC2, and HNRNPC were significantly downregulated in GBMs comparing with LGGs. We further validated these findings in additional datasets (Fig. s-1).

The m6a-related genes were dysregulated in gliomas from GSE16011 dataset. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. a, b The expression of 16 m6a-related genes
Identification of m6a-related lncRNAs in patients with pGBM
A total of 1248 (1901 probes) lncRNAs were identified from GSE16011 by using lncRNA expression profile mining [19, 20, 28]. We identified m6a-related lncRNAs by constructing m6a-lncRNA co-expression networks, and 494 lncRNAs (694 probes) were identified (Cor value 0.4, P ≤ 0.001). Based on these m6a-related lncRNA expression profiles, we conducted a principal component analysis (PCA) to investigate the difference between LGGs and pGBMs. It showed that LGGs and pGBMs were generally distributed in different directions (Fig. 2a).

Distinct m6a status was identified according to histologic grade, low-risk and high-risk groups. a LGG and pGBMs displayed different m6a status. b Low-risk and high-risk groups displayed different m6a status
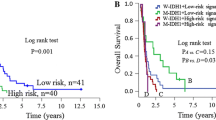
We collected 341 pGBM (GSE16011 150, CGGA microarray 108, CGGA RNA sequencing 83) patients from three datasets. The top five prognostic probes (P ≤ 0.01) are listed in Table 1. The m6a-related lncRNAs were MIR9-3HG, LINC00900, MIR155HG, and LINC00515. A five m6a-related-lncRNA signature was developed using a risk score method [23,24,25, 29]. Based on the median risk score, we divided the pGBMs into two groups (low-risk and high-risk groups). Patients in the high-risk group showed shorter overall survival (OS) than the low-risk group (median OS 7.80 vs. 11.04 months; P = 0.0025; Fig. 3a). These findings were further validated in two additional datasets (CGGA microarray, CGGA RNA sequencing) using the same β value, which showed similar results, respectively. Moreover, patients in the low-risk group showed longer progression-free survival (PFS) than the high-risk group (median PFS 406 vs. 271 days; P = 0.0070; Fig. 3b). Furthermore, two groups divided based on the median risk score displayed different m6a status (Fig. 2b).

Kaplan-Meier curves of OS and PFS among pGBM patients from different groups in three datasets. a The OS among pGBMs in different groups stratified by the signature in three datasets. b The PFS among pGBMs in different groups stratified by the signature in CGGA microarray dataset
Associations between the m6a-related lncRNAs and clinicopathologic features
We assessed the expression of the m6a-related lncRNAs in different grades. The results showed that LINC00900 and MIR155HG were increased with tumor grades, while MIR9-3HG and LINC00515 were decreased in three datasets (Fig. 4a). The expressions of LINC00900 and MIR155HG in the high-risk group were higher than those in the low-risk group, while MIR9-3HG and LINC00515 were lower in three datasets (Fig. 4b).

Expression of four m6a-related lncRNAs in different grades and groups. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. a The expression of four m6a-related lncRNAs in different grades. b The expression of four m6a-related lncRNAs in different groups
The clinicopathologic features were collected from the three databases. The patients in the high-risk group showed the features of older age, isocitrate dehydrogenase (IDH) wild-type, classical and mesenchymal TCGA subtype, and G3 CGGA subtype (Fig. 5a). These findings were further validated in two additional databases. The risk score was an independent factor based on the univariate and multivariate Cox regression analyses (P = 0.017, HR = 1.338) (Table s-1).

Distribution of lncRNA expression, clinicopathologic features, and functional annotation in GSE16011 dataset. a Heatmap of four m6a-related lncRNAs expression and clinicopathologic features in low-risk and high-risk groups. Rows represent corresponding genes, while columns indicate corresponding patients. b BP analysis of the differentially expressed genes in low-risk and high-risk groups
Functional annotation of the m6a-related lncRNAs
Functional annotation of the m6a-related lncRNAs was explored between two groups. The co-expressed genes were screened with the absolute value of Pearson correlation coefficient 0.5. Finally, we identified 1495 (895 positive, 600 negative) genes with co-expression networks in GSE16011. The biological process (BP) analysis was conducted using DAVID (The Database for Annotation, Visualization and Integrated Discovery). The results showed that differentially expressed genes between the two groups were enriched in immune response, apoptosis, cell adhesion, negative regulation of transcription, negative regulation of RNA metabolic process, and regulation of RNA metabolic process (Fig. 5b).
Discussion
A total of 355 pGBM patients were collected to explore the prognosis of m6a-related lncRNAs in three datasets (GSE16011, CGGA microarray, and CGGA RNA sequencing). We identified 494 lncRNAs (694 probes) according to the lncRNA-m6a co-expression networks. On the univariate Cox regression analysis, four m6a-related prognostic lncRNAs were screened. Using a risk score method, we created a risk score that could divide pGBMs into low- and high-risk groups based on the median value. The OS and PFS in the low-risk group were longer than those in high-risk group (Fig. 3). The multivariate Cox regression analysis showed that the risk score was an independent factor.
Mounting evidence suggests that m6a has critical roles in cancer pathogenesis. The effect of m6a on the mechanisms and functions of lncRNA methylation is unclear. Recently, it was reported that ALKBH5 could demethylate KCNK15-AS1 and regulate KCNK15-AS1-mediated cell migration and invasion [18]. The lncRNA XIST is highly methylated with at least 78 m6a residues. Knockdown of RBM15, RBM15B, or METTL3 impairs XIST-mediated gene silencing [8]. Moreover, lncRNA FOXM1-AS could facilitate the interaction of ALKBH5 with FOXM1 nascent transcripts and contribute to GSC maintenance [7]. The m6a may influence lncRNA splicing that might alter cancer progression [30].
Four m6a-related prognostic lncRNAs were identified in our study. MIR9-3HG and LINC00515 were protective genes, while LINC00900 and MIR155HG were risky genes. It was reported that MIR9-3HG could maintain neural stem cells in undifferentiated state by regulating NR2E1 [31] that plays an oncogenic role in prostate carcinogenesis [32]. MIR155HG is involved in colorectal cancer progression [33] and significantly correlated with the overall analysis of patients with kidney renal clear cell carcinoma [34]. Moreover, MIR155HG is upregulated in mesenchymal GBMs and blocking MIR155HG/miR-155 axis could inhibit mesenchymal transition in glioma [35, 36]. LINC00515 could enhance the autophagy and chemoresistance of melphalan-resistant myeloma by inhibiting miR-140-5p [37]. It showed that the expression of LINC00515 was decreased in cisplatin-resistant ovarian cancer cells [38].
Our study focuses on the m6a-related lncRNAs, and there are some limitations in our study. Our findings were explored based on bioinformatics analysis and needed to be further validated with additional datasets. The four lncRNAs may provide clues for discovering the mechanisms and functions of m6a for pGBM patients. Moreover, experimental research should be carried out on these lncRNAs.
In conclusion, the m6a-lncRNA co-expression networks were constructed and we identified four m6a-related prognostic lncRNAs for pGBMs. The expression of m6a status was significantly different between LGGs and pGBMs. Moreover, pGBM patients divided based on the median risk score also displayed a feature of different m6a status.
References
Yang P, Wang Y, Peng X, You G, Zhang W, Yan W, Bao Z, Wang Y, Qiu X, Jiang T (2013) Management and survival rates in patients with glioma in China (2004-2010): a retrospective study from a single-institution. J Neuro-Oncol 113:259–266
Yang Y, Hsu PJ, Chen YS, Yang YG (2018) Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28:616–624
Weng H, Huang H, Wu H et al (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22:191–205 e199
Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, Weitman S, Tomlinson GE, Rao MK, Kornblau SM, Bansal S (2014) WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28:1171–1174
Cui Q, Shi H, Ye P et al (2017) m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep 18:2622–2634
Liu J, Ren D, Du Z, Wang H, Zhang H, Jin Y (2018) m(6)A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem Biophys Res Commun 502:456–464
Zhang S, Zhao BS, Zhou A et al (2017) m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31:591–606 e596
Patil DP, Chen CK, Pickering BF et al (2016) m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369–373
Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, Chen J, Xu W, Zhong L, Sun X (2018) Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark 21:859–868
Bian EB, Li J, Xie YS, Zong G, Li J, Zhao B (2015) lncRNAs: new players in gliomas, with special emphasis on the interaction of lncRNAs with EZH2. J Cell Physiol 230:496–503
Sun Y, Wang Z, Zhou D (2013) Long non-coding RNAs as potential biomarkers and therapeutic targets for gliomas. Med Hypotheses 81:319–321
Zhang XQ, Leung GK (2014) Long non-coding RNAs in glioma: functional roles and clinical perspectives. Neurochem Int 77:78–85
Wang P, Ren Z, Sun P (2012) Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem 113:1868–1874
Wang Q, Zhang J, Liu Y, Zhang W, Zhou J, Duan R, Pu P, Kang C, Han L (2016) A novel cell cycle-associated lncRNA, HOXA11-AS, is transcribed from the 5-prime end of the HOXA transcript and is a biomarker of progression in glioma. Cancer Lett 373:251–259
Han Y, Wu Z, Wu T, Huang Y, Cheng Z, Li X, Sun T, Xie X, Zhou Y, du Z (2016) Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis 7:e2123
Ni W, Yao S, Zhou Y, Liu Y, Huang P, Zhou A, Liu J, Che L, Li J (2019) Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol Cancer 18:143
Wu Y, Yang X, Chen Z et al (2019) m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer 18:87
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, Liu D, Tian L, Yin J, Jiang K, Miao Y (2018) ALKBH5 inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem 48:838–846
Zhang X, Sun S, Pu JK, Tsang AC, Lee D, Man VO, Lui WM, Wong ST, Leung GK (2012) Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol Dis 48:1–8
Wang Y, Li Y, Yue M et al (2018) Publisher correction: N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci
Michelhaugh SK, Lipovich L, Blythe J, Jia H, Kapatos G, Bannon MJ (2011) Mining Affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J Neurochem 116:459–466
Johnson R (2012) Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol Dis 46:245–254
Cai J, Zhang W, Yang P, Wang Y, Li M, Zhang C, Wang Z, Hu H, Liu Y, Li Q, Wen J, Sun B, Wang X, Jiang T, Jiang C (2015) Identification of a 6-cytokine prognostic signature in patients with primary glioblastoma harboring M2 microglia/macrophage phenotype relevance. PLoS One 10:e0126022
Bao ZS, Li MY, Wang JY, Zhang CB, Wang HJ, Yan W, Liu YW, Zhang W, Chen L, Jiang T (2014) Prognostic value of a nine-gene signature in glioma patients based on mRNA expression profiling. CNS Neurosci Ther 20:112–118
Zhang CB, Zhu P, Yang P, Cai JQ, Wang ZL, Li QB, Bao ZS, Zhang W, Jiang T (2015) Identification of high risk anaplastic gliomas by a diagnostic and prognostic signature derived from mRNA expression profiling. Oncotarget 6:36643–36651
Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R (2004) Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med 350:1828–1837
Zhang W, Zhang J, Yan W, You G, Bao Z, Li S, Kang C, Jiang C, You Y, Zhang Y, Chen CC, Song SW, Jiang T (2013) Whole-genome microRNA expression profiling identifies a 5-microRNA signature as a prognostic biomarker in Chinese patients with primary glioblastoma multiforme. Cancer 119:814–824
Wang W, Yang F, Zhang L, Chen J, Zhao Z, Wang H, Wu F, Liang T, Yan X, Li J, Lan Q, Wang J, Zhao J (2016) LncRNA profile study reveals four-lncRNA signature associated with the prognosis of patients with anaplastic gliomas. Oncotarget 7:77225–77236
Wang W, Zhang L, Liu W, Zhu Q, Lan Q, Zhao J (2016) Antiplatelet agents for the secondary prevention of ischemic stroke or transient ischemic attack: a network meta-analysis. J Stroke Cerebrovasc Dis 25:1081–1089
Dai D, Wang H, Zhu L, Jin H, Wang X (2018) N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis 9:124
Nikkanen J, Landoni JC, Balboa D, Haugas M, Partanen J, Paetau A, Isohanni P, Brilhante V, Suomalainen A (2018) A complex genomic locus drives mtDNA replicase POLG expression to its disease-related nervous system regions. EMBO Mol Med 10:13–21
Jia L, Wu D, Wang Y, You W, Wang Z, Xiao L, Cai G, Xu Z, Zou C, Wang F, Teoh JY, Ng CF, Yu S, Chan FL (2018) Orphan nuclear receptor TLX contributes to androgen insensitivity in castration-resistant prostate cancer via its repression of androgen receptor transcription. Oncogene 37:3340–3355
Thiele JA, Hosek P, Kralovcova E et al (2018) lncRNAs in non-malignant tissue have prognostic value in colorectal cancer. Int J Mol Sci 19
Song J, Peng J, Zhu C, Bai G, Liu Y, Zhu J, Liu J (2018) Identification and validation of two novel prognostic lncRNAs in kidney renal clear cell carcinoma. Cell Physiol Biochem 48:2549–2562
Wu X, Wang Y, Yu T, Nie E, Hu Q, Wu W, Zhi T, Jiang K, Wang X, Lu X, Li H, Liu N, Zhang J, You Y (2017) Blocking MIR155HG/miR-155 axis inhibits mesenchymal transition in glioma. Neuro-Oncology 19:1195–1205
Balasubramaniyan V, Bhat KP (2017) Targeting MIR155HG in glioma: a novel approach. Neuro-Oncology 19:1152–1153
Lu D, Yang C, Zhang Z, Cong Y, Xiao M (2018) Knockdown of Linc00515 inhibits multiple myeloma autophagy and chemoresistance by upregulating miR-140-5p and downregulating ATG14. Cell Physiol Biochem 48:2517–2527
Zheng ZG, Xu H, Suo SS et al (2016) The essential role of H19 contributing to cisplatin resistance by regulating glutathione metabolism in high-grade serous ovarian cancer. Sci Rep 6:26093
Funding
This work was supported by grants from the Beijing Municipal Administration of Hospitals’ Mission Plan (SML20150501), the “13th Five-Year Plan” National Science and Technology supporting plan (2015BAI09B04), and the Foundation of Beijing Tiantan Hospital (2018-YQN-6).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Beijing Tiantan Hospital institutional review board (IRB).
Informed consent
Informed consent was obtained from each patient involved in our research.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wang, W., Li, J., Lin, F. et al. Identification of N6-methyladenosine-related lncRNAs for patients with primary glioblastoma. Neurosurg Rev 44, 463–470 (2021). https://doi.org/10.1007/s10143-020-01238-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-020-01238-x