
Abstract
A Staphylococcus aureus isolate (SA01) obtained from bloodstream infection exhibited a remarkable drug resistance profile. In this study, we report the draft genome sequence of S. aureus ST 5 SA01, a multidrug-resistant isolate, and analyzed the genes associated with drug resistance and virulence. The genome sketch of S. aureus ST5 SA01 was sequenced with Illumina and annotated using the Prokka software. Rapid Annotation Subsystem Technology (RAST) was used to verify the gene functions in the genome subsystems. The Comprehensive Antibiotic Resistance Database (CARD) and Virulence Factor Database (VFDB) were used in the analysis. The RAST indicated a contribution of 25 proteins to host adenine, fibronectin-binding protein A (FnbA), and biofilm formation as an intercellular polysaccharide adhesive system (PIA). The MLST indicated that S. aureus ST 5 SA01 belongs to ST5 (CC5). In silico analyses also showed an extensive repertoire of genes associated with toxins, such as LukGH leukocidin, enterotoxins, and superantigen staphylococcal classes (SSL). The 11 genes for antimicrobial resistance in S. aureus ST 5 SA01 showed similarity and identity above ≥ 99% with nucleotide sequences deposited in GenBank. Although studies on ST5 clones in Brazil are scarce, monitoring the clone of S. aureus ST 5 SA01 is essential, as it has become a problem in pediatrics in several countries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Staphylococcus aureus is an important Gram-positive bacterium, usually found in the human anterior nares and skin. As an opportunistic pathogen, it is involved in a range of clinical conditions, including sepsis, surgical wound infections, and severe pneumonia, and diabetic foot ulcers (Tong et al. 2015; Macedo et al. 2021; Buis et al. 2023). Moreover, this bacterium often acquires resistance to antibiotics routinely used in medical practice (Tong et al. 2015). The systemic dissemination of S. aureus is associated with various virulence determinants such as coagulase, lipases, adhesins, nucleases, hemolysin, and toxins (Cheung et al. 2021). The toxins harm many immune cells and are classified as gamma-hemolysin AB (HlgAB), gamma-hemolysin CB (HlgCB), leukotoxin GH (LukGH), leukotoxin ED (LukED), and Panton-Valentine leukocidin (PVL) (Ahmad-Mansour et al. 2021).
The α-, β-, γ-, and δ-hemolysin are responsible for erythrocyte lysis and may aggravate the clinical symptoms during tissue and bloodstream infections (Vandenesch et al. 2012; Duan et al. 2018). In addition, α-hemolysin is more relevant for the pathogenicity of S. aureus, as it is responsible for the formation of pores in the plasma membrane of various host cells. Thus, α-hemolysin can modulate multiple cellular processes, including excessive production of cytokines and trigger cell death (Virreira Winter et al. 2016).
In a previous study, S. aureus strains were isolated from bloodstream infections in São Luís, a city in Northeast Brazil that is part of the Legal Amazon area. One isolate, denominated S. aureus ST 5 SA01 showed resistance against many antibiotics (including clindamycin, erythromycin, gentamicin, rifampicin, and tetracycline) being classified as multidrug-resistant (MDR) (Monteiro et al. 2019). The drug resistance profile of this isolate encouraged the genome analysis of this strain since the sequence data obtained allow the further characterization of the mechanisms involved in drug resistance and virulence of bacterial pathogens (Kumburu et al. 2018; McManus et al. 2020; Jesus et al. 2022; Kumari et al. 2023).
This study reports the draft genome sequence of S. aureus ST 5 SA01 and comparative analyzes of genes associated with drug resistance and virulence using standard strains. The genomic data reported in this study are useful for future in silico analysis providing more insights about the spread of genes related with virulence and drug resistance.
Materials and methods
Sample
The bacterium isolate (S. aureus SA01) used in the study belong to the culture bank of the Laboratory of Applied Microbiology at the CEUMA University. It was previously isolated as part of other study with positive blood culture of patients hospitalized in Intensive Care Units (ICUs)(Monteiro et al. 2019). The bacterium is kept in glycerol stocks preserved at − 20 °C.
Antimicrobial susceptibility test and time-kill curve for oxacillin
The antimicrobial susceptibility profile of S. aureus ST 5 SA01 isolate was determined using AST #105 and GP-ID cards from the VITEK® 2 Compact system (BioMérieux, Marcy l’Etoile, France), according to the Clinical Laboratory Standards Institute (CLSI 2017). The susceptibility was evaluated for the following antibiotics: oxacillin, erythromycin, clindamycin, gentamicin, rifampicin, teicoplanin, vancomycin, trimethoprim/sulfamethoxazole, ciprofloxacin, and linezolid.
For the time-kill assay, the bacterial suspension (100 µL at 1 × 106 CFU/mL) was added to 900 µL of Muller Hinton broth (MHB) containing different oxacillin concentrations (32, 64, 128, and 256 μg/mL). At specific periods (0, 6, 12, 18, and 24 h), aliquots were tenfold diluted and plated for colony-forming unit (CFU) enumeration. The results were expressed as Log CFU/mL.
Genome sequencing, annotation, and Multilocus sequence typing analysis
The partial genome was sequenced using the Illumina MiSeq paired library approach and prepared by the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA). Pre-assembled genomic DNA sequences were annotated using the Prokka software tool (Seemann 2014). The sequence readings were assembled with the A5 software and processed for adapter cutting, quality filtering, and error correction to generate the contigs and scaffolds. In addition, the CAP3 software was used to improve scaffolding assembly, cut low-quality regions, and correct erroneous links between contigs.
Multilocus sequence typing analysis (MLST) was used to confirm the type of sequence (ST) and clonal complex. The sequences of seven housekeeping genes were analyzed: (i) arcC, (ii) aroE, (iii) glpF, (iv) gmk, (v) pta, (vi) tpi, and (vii) yqiL. The STs were obtained through an online web tool http://saureus.mlst.net) (Enright et al. 2000).
Partial analysis of Staphylococcus aureus SA01 genome
The annotated sequences of S. aureus ST 5 SA01 genome were analyzed using Rapid Annotation using Subsystem Technology (RAST), available at https://rast.nmpdr.org/rast.cgi (Aziz et al. 2008). Virulence factor genes were identified by comparison with the Virulence Factor Database (VFDB) (availabe at http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi) (Chen 2004). The Comprehensive Antibiotic Resistance Database (CARD) (available at https://card.mcmaster.ca) was used to determine the antibiotic resistance profile or resistance of S. aureus ST 5 SA01 (Alcock et al. 2019). A search for putative prophages in the genome of S. aureus ST 5 SA01 was performed with PHASTER (https://phaster.ca. (Arndt et al. 2016).
Mauve Contig Mover and BLAST Ring Image Generator (BRIG)
The Mauve Contig Mover was used to determine genome rearrangements after alignment. The genome S. aureus NCTC 8325 was selected as the reference in this analysis. The analysis was made for S. aureus SA01, S. aureus MS4, S. aureus Mu3, S. aureus Mu50, S. aureus N315, and S. aureus RF122. The comparison with other genomes was performed using Blast Ring Image Generator (BRIG) (Alikhan et al. 2011).
OrthoVenn and digital DNA-DNA hybridization analysis
A Venn diagram using OrthoVenn 2 (https://orthovenn2.bioinfotoolkits.net) (Xu et al. 2019) software allowed the visualization of genes encoding shared and unique proteins or pseudogenes between S. aureus ST 5 SA01 and S. aureus NCTC 8325 in each sequenced genome. Each lineage of S. aureus is represented by ovals of different colors with the number of groups of orthologs genes shared by the strains considered.
A phylogeny of the genome was generated using the TYGS server (http://tygs.dsmz.de.) (Meier-Kolthoff and Göker 2019). The generated genome sequence was used to determine the OGRI values about closely related Staphylococcus strains, including the digital DNA-DNA hybridization value (dDDH) calculated using GGDC web server formula two available at https://ggdc.dsmz.de/ggdc.php.
Results
Characterization of the Staphylococcus aureus SA01
The S. aureus ST 5 SA01 is resistant to oxacillin, erythromycin, gentamicin, and ciprofloxacin, and it is sensitive to teicoplanin, rifampicin, trimethoprim/sulfamethoxazole, vancomycin, and linezolid. In fact, S. aureus ST 5 SA01 is MDR isolate. The MLST analyses confirmed that it belongs to clonal complex 5 (CC5) and sequence type 5 (ST5).
The growth of the S. aureus ST 5 SA01 was evaluated in the presence of oxacillin at 32, 64, 128, and 256 μg/mL up to 24 h. The time-kill curve shows that S. aureus ST 5 SA01 did not grow in the presence of the highest concentrations tested (128 μg/mL and 256 μg/mL). The reductions in CFU counting for oxacillin at 64 μg/mL were 2.44, 4.59, 2.34, and 0.24 for incubation during 6, 12, 18, and 24 h, respectively. For oxacillin at 32 μg/mL, the reduction was only observed after 6 h of incubation (1.98) (Fig. 1).

Time-kill curve of Staphylococcus aureus SA01 grown in the presence of oxacillin at different concentrations (32, 64, 128, and 256 μg/mL)
Genome sequencing and annotation
The draft genome annotation data of S. aureus ST 5 SA01 are shown in (Table 1). Genomic sequencing data were deposited in the sequence read file database under Bio project prjna563016 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA563016), SAMN12661330 access biosample, Sequence Read Archive SRR10042834 under access (https://www.ncbi.nlm.nih.gov/search/all/?term=SRR10042834), and WGS was deposited with identification Access JANUHQ000000000 (https://www.ncbi.nlm.nih.gov/nuccore/JANUHQ000000000.1).
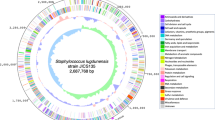
Identifying the sequences of coding genes (CDs) in the partial genome was 2687 CDs. The RAST analysis performed on several genes showed associations with several categories of subsystems (Fig. 2). However, this study highlights some genes that encode proteins associated with virulence and antibiotic resistance involved in iron capture. BLASTn compared these elements, and finally, the identities of the sequences were displayed.

Subsystem categories present non-genome of Staphylococcus aureus SA01 annotated by Rapid Annotation Subsystem Technology (RAST)
Rapid Annotation Subsystem Technology (RAST)
Rapid Annotation Subsystem Technology (RAST) analysis revealed the pathogenicity potential of S. aureus ST 5 SA01 showing 25 proteins involved in adhesion, such as sasA protein anchored in the predicted cell wall (LPXTG reason), aggregation factors A and B (ClfA and ClfB) and protein A of binding to fibronectin (FnbA). Genes related to intercellular polysaccharide adhesin (IAP) and elastin-binding protein were also identified (Table 2).
Genomic inferences in S. aureus ST 5 SA01 also indicated the presence of 28 genes related to the iron acquisition system (Supplementary Table 1). Of the proteins found, 18 are related to the transport and metabolism of the heme ring. Its amino acid sequences are highly similar to the sequences deposited from other strains of S. aureus.
Virulence Factor Database (VFDB)
The Virulence Factor Database (VFDB) system verified the broad spectrum of virulence genes. We expressed the results by comparing their absence and presence to seven lines of S. aureus Newman NCTC8325, N315, Mu3, MW2, JH1, and USA300 TCH1516 in the database (Fig. 3). The contribution of sa01 genome genes to virulence was quantified in 60.5% (n = 76) of VFDB, with most genes found (79%) related to enzymes, toxins, secretor systems, and adhesion. Comparatively, some genes for exotoxins detected in the genome of S. aureus ST 5 SA01 were the same as those seen in S. aureus NCTC 8325, S. aureus Mu3, S. aureus Mu50, and S. aureus N315.

Genes associated with virulence factors predicted in Staphylococcus aureus strains SA01 (1), Newman NCTC8325 (2), N315 (3), Mu3 (4), MW2 (5), JH1 (6), USA 300- TCH 1516 (7), and Mu50 (8). The blue network indicates the presence of virulence genes, while the empty network indicates their absence
Prediction of antibiotic resistance in Staphylococcus aureus SA01
The antimicrobial sensitivity profile of S. aureus ST 5 SA01 shows resistance to oxacillin, erythromycin, clindamycin, gentamicin, and ciprofloxacin, however sensitivity to teicoplanin, rifampicin, trimethoprim/sulfamethoxazole, vancomycin, and linezolid. The corroboration of these data in the genome of S. aureus ST 5 SA01 was possible to verify the presence of gyrA, parC, and mecA using the homologous protein model by the CARD software (Table 3). Other genes associated with antibiotic resistance were detected, such as efflux pumps, a superfamily of primary facilitators, and genes related to antibiotic target site modification (Table 3). The 11 genes showed similarity and identity above ≥ 99% with nucleotide.
Analysis of prophages
The presence of three distinct regions for bacteriophage sequences was verified through analysis of the genome of the S. aureus ST 5 SA01 scanned by PHASTER (Table 4, Supplementary Fig. 1). These were two incomplete sequences of 18.3 and 9.3 kb. In addition, the sequence of an entire region greater than 36.2 kb was verified, comprising 35 proteins, in which the most common phages were PHAGE_Staphy_phiMR25_NC_010808, PHAGE_Bacill_BtCS33_NC_018085, and PHAGE_Staphy_SPbeta_like_NC_029119.
In parallel to PHASTER, the VRprofile server was used to locate the homologs for gene ORFs conserved using an association of HMMer and BLASTp (using the value Ha 0.81). The analyses indicated nine regions with ORFs for prophages. The largest predicted region for phages was ORF 8, at 102 KB, with 149 ORFs and 44 hypothetical proteins for sequences traced in some genomes of S. aureus. In addition, the research indicated that many predicted sequences are related to prophages found in different strains of S. aureus. In addition, lines are associated with five types of enterotoxins or their precursors, such as enterotoxin Q, sec3-enterotoxin C1 precursor, entE-enterotoxin E precursor, speG-exotoxin G precursor, and seb-enterotoxin B, and also associated with hlgA precursor of hemolysin-gamma chain II.
Genomic analyses by MAUVE Contig mover and BLAST Ring Image Generator
In the analysis of genome similarities by MAUVE, preserved regions or blocks were observed, especially the total alignment of the seven strains of S. aureus, allowing the identification of 18 local choline blocks (LCBs), with between 6 and 9 LCBs presenting regions without a rearrangement of the homologous gene sequence (Fig. 4). The genome sketch of S. aureus ST 5 SA01 gives four LCBs in reverse orientation compared to the other isolates of S. aureus; however, the contigs of S. aureus NCTC 8325, MS4, Mu3, Mu50, N315, and RF122 had high similarity and homologous regions without rearrangements. This similarity is attributed to essential functions, such as virulence genes or determinants of antibiotic resistance, in the selected S. aureus strains.

Mauve comparison diagrams of the S. aureus NCTC8325, S. aureus MS4, S. aureus Mu3, S. aureus Mu50, S. aureus N315, and S. aureus RF122 genomes. Each colored region is a locally collinear block (LCB). The LCBs below the genome’s center line are in reverse complement orientation compared with the S. aureus NCTC8325 genome
Furthermore, MAUVE analyses showed synteny between the genome outline of S. aureus ST 5 SA01 with the complete genome of S. aureus NCTC 8325 (reference lineage) (Supplementary Fig. 2). MAUVE analysis revealed that a vast part of genetic information was conserved between the two strains.
In the analysis of BRIGs between S. aureus ST 5 SA01 and NCTC 8325, RF122, and Mu3, the region between 1600 and 1400 kbp showed abundant GC regions representing GC skew ( −) (Fig. 5). The region presented 100% similarity between SA01 and NCTC8325, differentiating them from the other strains of S. aureus.

Analysis of BRIGs among the strains of Staphylococcus aureus SA01, Mu50, Mu3, N315, RF122, and NCTC 8523. The innermost circle shows GC content. The cyan circle indicates the genome of NCTC 8523, and the lilac circle shows P Staphylococcus aureus SA01 genome. The white gaps indicate the absent sequences in the genomes
OrthoVenn and digital DNA-DNA hybridization analysis
Ortholog clusters in the S. aureus SA01, Mu50, Mu3, N315, and NCTC 8523 were analyzed using the OrthoVenn 2 software. The analysis of the strains indicated that they form 2873 orthologs clusters, which include 2211 central genome orthologs (Fig. 6A). S. aureus ST 5 SA01 presented five unique (exclusive) clusters with proteins with no defined function. There were 24,955 gene families containing sequences of the four gene strains S. aureus, of which 2211 (76.96%), 289 (10.06%), 143 (5.08%), 216 (7.52%), and 14 (0.49%) were shared by five, four, three, two, and one of these species, respectively. The five strains of S. aureus formed 2873 clusters, 680 orthologs clusters (containing at least two species), and 2193 clusters of single-copy genes.

Ortholog clusters in the Staphylococcus aureus strains SA01, Mu50, Mu3, N315, and NCTC 8523. A The table shows overlays identified by the OrthoVenn 2 analysis of S. aureus SA01, Mu50, Mu3, N315, and NCTC 8523. B The Venn diagram shows the distribution of ortholog groups shared among the S. aureus SA01, Mu50, Mu3, N315, and NCTC 8523
Figure 7 shows the functional classification of the proteins belonging to clusters the 2211 clusters shared by the five strains of S. aureus. Most proteins were classified as involved in biological, metabolic, and cellular metabolic processes. In this case, the biological processes represent a specific objective for which the organism is genetically programmed, such as cell division. On the other hand, metabolic processes involve chemical reactions and their pathways, through which living organisms transform chemicals; while, cellular metabolic processes are chemical reactions and their pathways that occur within the cell to transform chemicals (Fig. 7A).

Functional classification of proteins belonging to clusters in common among strains of S. aureus: SA01, NCTC 8325, Mu50, Mu3, N315. A Metabolic functions; B molecular functions; C cellular localization
When analyzed from the point of view of molecular functions (Fig. 7B), 16% of the proteins present hydrolase activity, which is a class of enzymes responsible for breaking a chemical bond and dividing a large molecule into two smaller ones; 15% of the proteins are involved in a molecular process involving the action or activity of a gene product, and 13% are responsible for the movement of substances inside, outside, and between cells.
Regarding the cellular distribution, most proteins were classified as part of the cell and cell membrane (Fig. 7C).
The results of the TYGS database indicated high similarity between S. aureus ST 5 SA01 and S. aureus N315, with 98.7% for dDhD; S. aureus ST 5 SA01 and S. aureus Mu50, with 98.1% for dDhD. However, we observed a value of 89.9% for DDDH between S. aureus ST 5 SA01 and NCTC 8325 (reference lineage), with a difference of 0.05% in the content of G + C.
Phenotypic and genotypic differences and clonal relationships are supported by genomic relationships (ANI and dDDH). The phylogenetic analysis of the genome sequence sketch of S. aureus ST 5 SA01 with other Staphylococcus strains revealed a cluster of S. aureus ST 5 SA01 with S. aureus N315 and S. aureus Mu50 are shown in Fig. 8. According to these results, genome analysis confirmed that S. aureus ST 5 SA01 actually belongs to S. aureus sub. aureus with ANI values of 98.76%; therefore, S. aureus sub. aureus N315, S. aureus sub. aureus Mu50, and S. aureus sub. aureus NCTC 8325 have ANI values of 98.81, 98.72, and 99.75%, respectively (Supplementary Table 2).

The phylogenetic tree of Staphylococcus aureus SA01 and related Staphylococcus strains available in the TYGS database. The tree inferred with FastME 2.1.6.1 from the Genome BLAST Distance Phylogeny (GBDP) method shows the distances calculated from genome sequences. The lengths of the branches are scaled in terms of the distance formula d5 GBDP. The numbers above the branches are GBDP pseudo-bootstrap support values of 100 repetitions, with average agency support of 95.5%
Discussion
S. aureus ST5 SA01 strains have become a problem in hospital environments and are spreading on several continents, and studies showing its dispersion are scarce in Brazil. In previous studies, it was observed that strains of S. aureus ST5 showed resistance rates against many antibiotics, including clindamycin, erythromycin, gentamicin, rifampicin, and tetracycline (Kim et al. 2019; Monteiro et al. 2019). The success of S. aureus ST5 inside and outside a hospital environment is attributed to the ability of these lineages to acquire moving elements, such as transposons and prophages, which contain genes that encode virulence and resistance factors (Monecke et al. 2011).
The time-kill curve assay can be used to evaluate the bactericidal activity of antimicrobials against the most diverse microorganisms, among them S. aureus SA01, allowing observation of the interaction dynamics between antimicrobial organisms (Di Pilato et al. 2020). Treatment with concentrations of 32 and 64 μg/mL of oxacillin did not inhibit the cellular viability of S. aureus SA01. At the beginning of the exponential phase, the cells added in medium without oxacillin (control) showed rapid growth, and the treatment with a concentration of 32 μg/mL had the same tendency, demonstrating a latency phase observed compared to the rule, making clear the adaptation stage of S. aureus ST 5 SA01 to a new environment. Through macromolecular repair and cell growth synthesis through DNA replication and thus corroborating other studies (Zhou et al. 2017). According to the antimicrobial resistance profile of S. aureus SA01, the use of combination therapy has been a beneficial strategy for the treatment of certain infections with tolerant microorganisms or in a biofilm, such as those associated with devices such as catheters and prostheses, so the critical role of the in vitro synergism test stands out (Belley et al. 2008; Watson et al. 2020; Rieg et al. 2020).
Many of the factors found in the genome of S. aureus ST 5 SA01 are genes that encode proteins that may play an individual or additive role in the pathogenesis of bacteria in host tissues (Table 2). Adhesion to host cells is mediated by proteins associated with the bacterial cell wall, called microbial surface components that recognize adhesive matrix molecules (MSCRAMMs) (Alli et al. 2015). The binding protein—fibronectin (FnbA) found in the genome S. aureus SA01—is responsible for promoting tissue adhering to the extracellular matrix. In S. aureus, FnbA is essential in the early stage of infection and has been implicated in infectious endocarditis and osteomyelitis (Murai et al. 2016; Soltani et al. 2019).
In this study, cIfA and clfB, found in the genome of S. aureus SA01, are responsible for the transcription of aggregation factors A and B, respectively. The CIfA, which is present in all phases of bacterial growth, can connect to the complement factor system and facilitate immune system evasion. ClfB, present in the exponential phase of bacterial growth, can bind to fibrin and cytokeratin (Haim et al. 2010; Liesenborghs et al. 2018). Previously, it was reported that aggregation factors A and B (ClfA and ClfB) are involved in the interactions of S. aureus with host-specific receptors, increasing the pathogen persistence at the infection site (Bonar et al. 2015; Hodille et al. 2017). In addition, the adhesivity and the ability of biofilm formation of S. aureus to abiotic surfaces are related to the production of polysaccharide intercellular adhesive (IAP) (You et al. 2014). In its genome, the S. aureus SA01 strain contains genes organized in the icaABCD operon, responsible for producing PIA, another host gene with regulatory function. The expression of icaD and icaA is associated with the initial phase of biofilm formation (Kot et al. 2018).
S. aureus produces a phospholipase specific to sphingomyelin called beta-hemolysin (HLB). HLB and gamma hemolysin (HLG) induce cellular lysis and are secreted by most isolates of S. aureus concerning chronic infections of human skin, systemic infections, and septic arthritis (Katayama et al. 2013; Soltani et al. 2019). We found that hlgA, hlgB, and hlgC in the genome of S. aureus ST 5 SA01 encode for hemolysin gamma. Interestingly, all strains of S. aureus used for comparison carried the genes for hemolysin gamma. HLG influences the pathogenicity of S. aureus and aggravates the symptoms of patients by being a potent leukotoxic and hemolytic stimulator (Koymans et al. 2015; Liesenborghs et al. 2018). After analysis by RAST BLASTn, the proteins LukG, and LukH presented 100% identity with other proteins from the GenBank database (Table 2).
The LukGH system is one of the most potent two-component leukocidin systems (Trstenjak et al. 2020). It can be compared with the strength of LukSF-PV in damaging human phagocytic cells, with action similar to PVL, contributing to immune evasion and cell lysis (Yanai et al. 2014). LukGH genes are an essential part of the Genome of S. aureus, while the lukSF/PVL gene, encoded by prophages, is expressed by only 5–10% of the clinical isolates of S. aureus (Vandenesch et al. 2012). After genomic analysis of S. aureus ST 5 SA01 proteins, staphylococcal superantigen classes (SSL) (SSL1, SSL2, SSL3, SSL4, SSL5, SSL7, SSL8, SSL9, and SSL10) were found. The SSL protein class is one of the prominent proteins secreted by S. aureus strains related to the invasion and colonization of host tissues (Thammavongsa et al. 2015; Lewis and Surewaard 2018; Bretl et al. 2019). SSLs are immune avoidance molecules that directly interfere with a range of innate and adaptive immune defense responses, which may be associated with the blockade, degradation, cell lysis, and modulation of immune function (Koymans et al. 2015). The genes of the SSL class detected in the genome of S. aureus ST 5 SA01 have structural similarities and distinct roles. These genes are located in the nucleus of the genome, corroborating other studies (Katayama et al. 2013; Zhao et al. 2018).
S. aureus is a species of bacteria that needs iron for critical biochemical reactions. This requirement is linked to the hemolytic capacity of the bacterium, leading to the depletion of host erythrocytes, mainly associated with infectious conditions such as sepsis (Roetzer et al. 2016). Two transport systems regulated by ISD and HTS involved in iron heme acquisition were detected in S. aureus SA01. These genes express proteins anchored to the cell wall, such as IsdA, IsdB, IsdC, and IsdH, and membrane transporters, such as IsdDEF (Liu et al. 2008). The ISD components allow S. aureus to extract the heme from hemoglobin (Hb), transport it to the bacterial cytoplasm and release iron from the porphyrin ring, thus, contributing to microbial pathogenesis (Conroy et al. 2019; Gianquinto et al. 2019; Mikkelsen et al. 2020). However, the role of these systems in the physiology of S. aureus is controversial (Mason and Skaar 2009; Sheldon and Heinrichs 2015).
The percentage of enterotoxin-related genes in S. aureus varies according to the strain and the types of phages found in the genome. In S. aureus SA01, genes related to staphylococcal enterotoxins, such as sec, yent1, yent2, selk, selm, seln, and seal, were found. These represented a rate of 35% (7/20) of the same genes detected in the Mu50 and N315 strains, at a rate of 50% (Kuroda et al. 2001; Collery et al. 2009; Arabestani et al. 2018) (Fig. 3 A–C, Supplementary Table 2).
The resistance of S. aureus ST 5 SA01 to ciprofloxacin (a fluoroquinolone) is due to mutations present in gyrA, gyrB, parE, and parC that encode the subunits of DNA gyrase and topoisomerase IV, the primary sites of action for these drugs. These mutations occur mainly in quinolone resistance determinant regions (QRDRs) (Röderova et al. 2017). Earlier studies have shown that ciprofloxacin-resistant S. aureus isolates showed gyrA and parC mutations, causing a reduction in quinoline binding affinity to enzymes gyrase and topoisomerase (Fuzi 2016; de Oliveira et al. 2019).
However, other mechanisms may be associated with quinolone resistance, such as changes in antibiotic flow systems; changes in the target action site of these drugs were also seen in the genome of S. aureus ST 5 SA01 (Ardebili et al. 2014). The presence of mecA, which encodes a penicillin-binding protein, altered in the genome of S. aureus ST 5 SA01 is closely related to oxacillin resistance. Resistance to methicillin and oxacillin occurs due to a mutation that leads to a change in penicillin-binding protein 2 (PBP2) that confers resistance to S. aureus SA01. The molecular pattern observed for S. aureus ST 5 SA01 is similar to that found for other strains of S. aureus, such as methicillin-resistant S. aureus (MRSA), isolated from the bloodstream (Chen et al. 2014).
S. aureus strains classified as ST5 are essential pathogens of community-associated methicillin-resistant S. aureus (CA-MRSA), epidemiologically relevant with worldwide distribution, which has been associated with severe invasive diseases in humans, especially in children (Sola et al. 2012; Rokney et al. 2019). Genome sketch analyses of S. aureus ST 5 SA01 indicated virulence factors relevant to bacterial pathogenesis in humans associated with phages. In addition, the contribution of phages is relevant to the pathogenesis with the acquisition of leukocidin genes and other aggression factors (Coombs et al. 2020).
The region of similarity between S. aureus ST 5 SA01 and S. aureus NCTC 8325, located in the ring (BRIG), about the part of the slope GC ( −), between 1600 and 1400 kbp, is represented by the imbalance of nucleotide concentration as the frequency of the four DNA bases, which was not observed in the other lines (Lobry 1996). This similarity with the S. aureus NCTC 8325 lineage, dated as a strain of S. aureus containing a high concentration of nucleotide exchange (mutation) in the gene composition, reflects selective pressure during the host infectious process (Kumburu et al. 2018). However, the presence of mobile genetic elements in the S. aureus ST 5 SA01 strains assumes the necessary advantages, capable of promoting the cellular adaptation of microorganisms to infected hosts, where this characteristic is due to the performance potential of strains of S. aureus in the horizontal transfer of genes associated with IGs (Moon et al. 2016; Turner et al. 2019; Kläui et al. 2019).
The high similarity between the sequences of strains S. aureus SA01, S. aureus N315, S. aureus Mu50, and S. aureus NCTC 8325 obtained by TYGS is congruent with the results obtained by the OrthoVenn Web server, and S. aureus strains may perform similar biological functions (Liang et al. 2019; Chamon et al. 2020). These proteins found in the genome of S. aureus ST 5 SA01 are related to necrosis, hemolysins GO-0001906, regulation of symbiosis in the host GO-0009405, enterotoxins GO-0090729 and go-07155 adhering to being a characteristic of the species, and may be related to a highly virulent profile, allowing the prevalence of this lineage in our environment, previous corroborating studies (School et al. 2016). In addition, the strains of S. aureus SA01, S. aureus N315, and S. aureus Mu50 belong to the sequence type group, classified as ST5 (CC5), unlike the S. aureus NCTC 8325 lineage, classified as ST8 (CC8).
Although S. aureus N315 is a methicillin-resistant S. aureus (MRSA) strain that was isolated in 1982, and Mu50 is a vancomycin-resistant S. aureus (VRSA) lineage that was isolated in 1997, its genomes were characterized in 2001 (Kuroda et al. 2001; Roetzer et al. 2016). Interestingly, they contain two copies of Tn554 in a site-specific transposon related to resistance to macrolide-lincosamide-streptogramin B (Kuroda et al. 2001). Coincidentally, none of the three strains of S. aureus—SA01, N315, and Mu50—carries lukS-PV, and lukF-PV encoding for PVL, although some reports indicate that ST5 can express this leukocidin (Rokney et al. 2019).
Conclusions
Here, the findings of the genomic characterization of S. aureus ST 5 SA01, a lineage belonging to the ST5 group, demonstrate the extensive repertoire of genes involved in antimicrobial resistance and virulence, such as leukocidin GH, a potent pore-forming toxin in human leukocytes. Although studies on ST5 clones in Brazil are scarce, it is essential to monitor this clone, as it has become a problem in pediatrics in some countries.
Data availability
All data generated or analyzed during this study are included in this article [and its supplementary information files].
References
Ahmad-Mansour N, Loubet P, Pouget C, et al (2021) Staphylococcus aureus toxins: an update on their pathogenic properties and potential treatments. Toxins (Basel) 13:. https://doi.org/10.3390/TOXINS13100677
Alcock BP, Raphenya AR, Lau TTY et al (2019) CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz935
Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. https://doi.org/10.1186/1471-2164-12-402
Alli OT, Ogbolu D, Shittu A et al (2015) Association of virulence genes with mecA gene in Staphylococcus aureus isolates from Tertiary Hospitals in Nigeria. Indian J Pathol Microbiol 58:464. https://doi.org/10.4103/0377-4929.168875
Arabestani MR, Rastiyani S, Alikhani MY, Mousavi SF (2018) The relationship between prevalence of antibiotics resistance and virulence factors genes of MRSA and MSSA strains isolated from clinical samples, West Iran. Oman Med J 33:134–140. https://doi.org/10.5001/omj.2018.25
Ardebili A, Talebi M, Azimi L, Rastegar Lari A (2014) Effect of efflux pump inhibitor carbonyl cyanide 3-chlorophenylhydrazone on the minimum inhibitory concentration of ciprofloxacin in Acinetobacter baumannii clinical isolates. Jundishapur J Microbiol 7: https://doi.org/10.5812/jjm.8691
Arndt D, Grant JR, Marcu A et al (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. https://doi.org/10.1093/nar/gkw387
Aziz RK, Bartels D, Best AA et al (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. https://doi.org/10.1186/1471-2164-9-75
Belley A, Neesham-Grenon E, Arhin FF et al (2008) Assessment by time-kill methodology of the synergistic effects of oritavancin in combination with other antimicrobial agents against Staphylococcus aureus. Antimicrob Agents Chemother 52:3820–3822. https://doi.org/10.1128/AAC.00361-08
Bonar E, Wójcik I, Wladyka B (2015) Proteomics in studies of Staphylococcus aureus virulence. Acta Biochim Pol 62:367–381. https://doi.org/10.18388/abp.2015_1083
Bretl DJ, Elfessi A, Watkins H, Schwan WR (2019) Regulation of the staphylococcal superantigen-like protein 1 gene of community-associated methicillin-resistant Staphylococcus aureus in murine abscesses. Toxins (basel) 11:391. https://doi.org/10.3390/toxins11070391
Buis DTP, van der Vaart TW, Prins JM et al (2023) Comparative effectiveness of β-lactams for empirical treatment of methicillin-susceptible Staphylococcus aureus bacteraemia: a prospective cohort study. J Antimicrob Chemother. https://doi.org/10.1093/JAC/DKAD057
Chamon RC, Marques LM, Timenetsky J et al (2020) Genome sequence of a highly virulent pvl-positive vancomycin-intermediate-resistant Staphylococcus aureus sequence type 30. Curr Genomics 21:128–137. https://doi.org/10.2174/1389202921666200327105756
Chen F-J, Wang C-H, Chen C-Y et al (2014) Role of the mecA gene in oxacillin resistance in a Staphylococcus aureus clinical strain with a pvl-positive ST59 genetic background. Antimicrob Agents Chemother 58:1047–1054. https://doi.org/10.1128/AAC.02045-13
Chen L, YJYJYZSLSYJQ (2004) VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33:D325–D328. https://doi.org/10.1093/nar/gki008
Cheung GYC, Bae JS, Otto M (2021) Pathogenicity and virulence of Staphylococcus aureus. Virulence 12:547–569. https://doi.org/10.1080/21505594.2021.1878688
CLSI (2017) Performance standards for antimicrobial susceptibility testing, 27th edn. Clinical and Laboratory Standards Institute, Wayne, PA
Collery MM, Smyth DS, Tumilty JJG et al (2009) Associations between enterotoxin gene cluster types egc1, egc2 and egc3, agr types, enterotoxin and enterotoxin-like gene profiles, and molecular typing characteristics of human nasal carriage and animal isolates of Staphylococcus aureus. J Med Microbiol 58:13–25. https://doi.org/10.1099/jmm.0.005215-0
Conroy BS, Grigg JC, Kolesnikov M et al (2019) Staphylococcus aureus heme and siderophore-iron acquisition pathways. Biometals 32:409–424. https://doi.org/10.1007/S10534-019-00188-2
Coombs GW, Baines SL, Howden BP, et al (2020) Diversity of bacteriophages encoding Panton-Valentine leukocidin in temporally and geographically related Staphylococcus aureus. PLoS One 15:e0228676. https://doi.org/10.1371/journal.pone.0228676
de Monteiro A, S, Pinto BLS, Monteiro J de M, et al (2019) Phylogenetic and molecular profile of Staphylococcus aureus isolated from bloodstream infections in northeast Brazil. Microorganisms 7:210. https://doi.org/10.3390/microorganisms7070210
de Oliveira TLR, Cavalcante FS, Chamon RC et al (2019) Genetic mutations in the quinolone resistance-determining region are related to changes in the epidemiological profile of methicillin-resistant Staphylococcus aureus isolates. J Glob Antimicrob Resist 19:236–240. https://doi.org/10.1016/j.jgar.2019.05.026
Di Pilato V, Ceccherini F, Sennati S, et al (2020) In vitro time-kill kinetics of dalbavancin against Staphylococcus spp. biofilms over prolonged exposure times. Diagn Microbiol Infect Dis 96:114901. https://doi.org/10.1016/j.diagmicrobio.2019.114901
Duan J, Li M, Hao Z et al (2018) Subinhibitory concentrations of resveratrol reduce alpha-hemolysin production in Staphylococcus aureus isolates by downregulating saeRS. Emerg Microbes Infect 7:1–10. https://doi.org/10.1038/s41426-018-0142-x
Enright MC, Day NPJ, Davies CE et al (2000) Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 38:1008–1015. https://doi.org/10.1128/JCM.38.3.1008-1015.2000
Fuzi M (2016) Dissimilar fitness associated with resistance to fluoroquinolones influences clonal dynamics of various multiresistant bacteria. Front Microbiol 7. https://doi.org/10.3389/fmicb.2016.01017
Gianquinto E, Moscetti I, De Bei O et al (2019) Interaction of human hemoglobin and semi-hemoglobins with the Staphylococcus aureus hemophore IsdB: a kinetic and mechanistic insight. Sci Rep 9:18629. https://doi.org/10.1038/s41598-019-54970-w
Haim M, Trost A, Maier CJ et al (2010) Cytokeratin 8 interacts with clumping factor B: a new possible virulence factor target. Microbiology (n y) 156:3710–3721. https://doi.org/10.1099/mic.0.034413-0
Hodille E, Rose W, Diep BA et al (2017) The role of antibiotics in modulating virulence in Staphylococcus aureus. Clin Microbiol Rev 30:887–917. https://doi.org/10.1128/CMR.00120-16
Jesus HNR, Ramos JN, Rocha DJPG, et al (2022) The pan-genome of the emerging multidrug-resistant pathogen Corynebacterium striatum. Functional & Integrative Genomics 2022 23:1 23:1–15. https://doi.org/10.1007/S10142-022-00932-X
Katayama Y, Baba T, Sekine M et al (2013) Beta-hemolysin promotes skin colonization by Staphylococcus aureus. J Bacteriol 195:1194–1203. https://doi.org/10.1128/JB.01786-12
Kim C-J, Song K-H, Choe PG et al (2019) The microbiological characteristics of Staphylococcus aureus isolated from patients with native valve infective endocarditis. Virulence 10:948–956. https://doi.org/10.1080/21505594.2019.1685631
Kläui AJ, Boss R, Graber HU (2019) Characterization and comparative analysis of the Staphylococcus aureus genomic island v Saβ: an in silico approach. J Bacteriol 201. https://doi.org/10.1128/JB.00777-18
Kot B, Sytykiewicz H, Sprawka I (2018) Expression of the biofilm-associated genes in methicillin-resistant Staphylococcus aureus in biofilm and planktonic conditions. Int J Mol Sci 19:3487. https://doi.org/10.3390/ijms19113487
Koymans KJ, Vrieling M, Gorham RD, van Strijp JAG (2015) Staphylococcal immune evasion proteins: structure, function, and host adaptation. pp 441–489
Kumari K, Sharma PK, Shikha S, Singh RP (2023) Molecular characterization and in-depth genome analysis of Enterobacter sp. S-16. Funct Integr Genomics 23:245. https://doi.org/10.1007/S10142-023-01161-6/TABLES/3
Kumburu HH, Sonda T, Leekitcharoenphon P et al (2018) Hospital epidemiology of methicillin-resistant Staphylococcus aureus in a Tertiary Care Hospital in Moshi, Tanzania, as determined by whole genome sequencing. Biomed Res Int 2018:1–12. https://doi.org/10.1155/2018/2087693
Kuroda M, Ohta T, Uchiyama I et al (2001) Whole genome sequencing of meticillin-resistant Staphylococcus aureus. The Lancet 357:1225–1240. https://doi.org/10.1016/S0140-6736(00)04403-2
Lewis ML, Surewaard BGJ (2018) Neutrophil evasion strategies by Streptococcus pneumoniae and Staphylococcus aureus. Cell Tissue Res 371:489–503. https://doi.org/10.1007/s00441-017-2737-2
Liang C-Y, Yang C-H, Lai C-H et al (2019) Comparative genomics of 86 whole-genome sequences in the six species of the elizabethkingia genus reveals intraspecific and interspecific divergence. Sci Rep 9:19167. https://doi.org/10.1038/s41598-019-55795-3
Liesenborghs L, Verhamme P, Vanassche T (2018) Staphylococcus aureus, master manipulator of the human hemostatic system. J Thromb Haemost 16:441–454. https://doi.org/10.1111/jth.13928
Liu M, Tanaka WN, Zhu H et al (2008) Direct hemin transfer from IsdA to IsdC in the iron-regulated surface determinant (Isd) heme acquisition system of Staphylococcus aureus. J Biol Chem 283:6668–6676. https://doi.org/10.1074/jbc.M708372200
Lobry JR (1996) Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol 13:660–665. https://doi.org/10.1093/OXFORDJOURNALS.MOLBEV.A025626
Macedo GHRV, Costa GDE, Oliveira ER et al (2021) Interplay between eskape pathogens and immunity in skin infections: an overview of the major determinants of virulence and antibiotic resistance. Pathogens 10:1–34. https://doi.org/10.3390/pathogens10020148
Mason WJ, Skaar EP (2009) Assessing the contribution of heme-iron acquisition to Staphylococcus aureus pneumonia using computed tomography. PLoS One 4:e6668. https://doi.org/10.1371/journal.pone.0006668
McManus BA, Daly B, Polyzois I, et al (2020) Comparative microbiological and whole-genome analysis of Staphylococcus aureus populations in the oro-nasal cavities, skin and diabetic foot ulcers of patients with type 2 diabetes reveals a possible oro-nasal reservoir for ulcer infection. Front Microbiol 11. https://doi.org/10.3389/fmicb.2020.00748
Meier-Kolthoff JP, Göker M (2019) TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun 10:2182. https://doi.org/10.1038/s41467-019-10210-3
Mikkelsen JH, Runager K, Andersen CBF (2020) The human protein haptoglobin inhibits IsdH-mediated heme-sequestering by Staphylococcus aureus. J Biol Chem 295:1781–1791. https://doi.org/10.1074/jbc.RA119.011612
Monecke S, Coombs G, Shore AC et al (2011) A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6:e17936. https://doi.org/10.1371/journal.pone.0017936
Moon BY, Park JY, Robinson DA, et al (2016) Mobilization of genomic islands of Staphylococcus aureus by temperate bacteriophage. PLoS One 11:e0151409. https://doi.org/10.1371/journal.pone.0151409
Murai M, Moriyama H, Hata E et al (2016) Variation and association of fibronectin-binding protein genes fnbA and fnbB in Staphylococcus aureus Japanese isolates. Microbiol Immunol 60:312–325. https://doi.org/10.1111/1348-0421.12377
Rieg S, Ernst A, Peyerl-Hoffmann G et al (2020) Combination therapy with rifampicin or fosfomycin in patients with Staphylococcus aureus bloodstream infection at high risk for complications or relapse: results of a large prospective observational cohort. J Antimicrob Chemother. https://doi.org/10.1093/jac/dkaa144
Röderova M, Halova D, Papousek I, et al (2017) Characteristics of quinolone resistance in Escherichia coli isolates from humans, animals, and the environment in the Czech Republic. Front Microbiol 7. https://doi.org/10.3389/fmicb.2016.02147
Roetzer A, Haller G, Beyerly J et al (2016) Genotypic and phenotypic analysis of clinical isolates of Staphylococcus aureus revealed production patterns and hemolytic potentials unlinked to gene profiles and source. BMC Microbiol 16:13. https://doi.org/10.1186/s12866-016-0630-x
Rokney A, Baum M, Ben-Shimol S et al (2019) Dissemination of the methicillin-resistant Staphylococcus aureus pediatric clone (ST5-T002-IV-PVL+) as a major cause of community-associated staphylococcal infections in bedouin children, Southern Israel. Pediatric Infect Disease J 38:230–235. https://doi.org/10.1097/INF.0000000000002126
School K, Marklevitz J, Schram WK, Harris LK (2016) Predictive characterization of hypothetical proteins in Staphylococcus aureus NCTC 8325. Bioinformation 12:209–220. https://doi.org/10.6026/97320630012209
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Sheldon JR, Heinrichs DE (2015) Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol Rev 39:592–630. https://doi.org/10.1093/femsre/fuv009
Sola C, Paganini H, Egea AL, et al (2012) Spread of epidemic MRSA-ST5-IV clone encoding PVL as a major cause of community onset staphylococcal infections in Argentinean children. PLoS One 7:e30487. https://doi.org/10.1371/journal.pone.0030487
Soltani E, Farrokhi E, Zamanzad B et al (2019) Prevalence and distribution of adhesins and the expression of fibronectin-binding protein (FnbA and FnbB) among Staphylococcus aureus isolates from Shahrekord Hospitals. BMC Res Notes 12:49. https://doi.org/10.1186/s13104-019-4055-0
Thammavongsa V, Kim HK, Missiakas D, Schneewind O (2015) Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. https://doi.org/10.1038/nrmicro3521
Tong SYC, Davis JS, Eichenberger E et al (2015) Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. https://doi.org/10.1128/CMR.00134-14
Trstenjak N, Milić D, Graewert MA et al (2020) Molecular mechanism of leukocidin GH–integrin CD11b/CD18 recognition and species specificity. Proc Natl Acad Sci 117:317–327. https://doi.org/10.1073/pnas.1913690116
Turner NA, Sharma-Kuinkel BK, Maskarinec SA et al (2019) Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol 17:203–218. https://doi.org/10.1038/s41579-018-0147-4
Vandenesch F, Lina G, Henry T (2012) Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front Cell Infect Microbiol 2. https://doi.org/10.3389/fcimb.2012.00012
Virreira Winter S, Zychlinsky A, Bardoel BW (2016) Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus α-hemolysin-mediated toxicity. Sci Rep 6:24242. https://doi.org/10.1038/srep24242
Watson A, Sauve K, Cassino C, Schuch R (2020) Exebacase demonstrates in vitro synergy with a broad range of antibiotics against both methicillin-resistant and methicillin-susceptible Staphylococcus aureus. Antimicrob Agents Chemother 64. https://doi.org/10.1128/AAC.01885-19
Xu L, Dong Z, Fang L et al (2019) OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res 47:W52–W58. https://doi.org/10.1093/nar/gkz333
Yanai M, Rocha MA, Matolek AZ, et al (2014) Separately or combined, LukG/LukH is functionally unique compared to other staphylococcal bicomponent leukotoxins. PLoS One 9:e89308. https://doi.org/10.1371/journal.pone.0089308
You Y, Xue T, Cao L et al (2014) Staphylococcus aureus glucose-induced biofilm accessory proteins, GbaAB, influence biofilm formation in a PIA-dependent manner. Int J Med Microbiol 304:603–612. https://doi.org/10.1016/j.ijmm.2014.04.003
Zhao Y, van Kessel KPM, de Haas CJC, et al (2018) Staphylococcal superantigen-like protein 13 activates neutrophils via formyl peptide receptor 2. Cell Microbiol 20:e12941. https://doi.org/10.1111/cmi.12941
Zhou T, Li Z, Kang O-H et al (2017) Antimicrobial activity and synergism of ursolic acid 3-O-α-L-arabinopyranoside with oxacillin against methicillin-resistant Staphylococcus aureus. Int J Mol Med 40:1285–1293. https://doi.org/10.3892/ijmm.2017.3099
Acknowledgements
The authors thank Hélio Euclides S. dos Santos and Marinaldo for technical assistance.
Funding
This study was funded by Fundação de Amparo à Pesquisa e o Desenvolvimento Científico e Tecnológico do Maranhão (FAPEMA, BM-05512/19; INFRA-02032/21; POS-GRAD-02460/21).
Author information
Authors and Affiliations
Contributions
Investigation: Romulo Ferreira, Douglas Santos Silva, Joveliane Monteiro, Gabriella Ferreira, Karinny Silva, Maria Raimunda Silva, and Letícia Oliveira; writing—original draft: Romulo Ferreira; writing—review and editing: Luís Claudio Nascimento da Silva and Andrea Monteiro.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Our study did not involve human or human tissue samples. The bacterium isolate used in the study was previously isolated as part of other published study and now is part of our Microbiological collection.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ferreira, R.M., dos Santos Silva, D.H., Silva, K.F. et al. Draft genome sequence of Staphylococcus aureus sequence type 5 SA01 isolated from bloodstream infection and comparative analysis with reference strains. Funct Integr Genomics 23, 288 (2023). https://doi.org/10.1007/s10142-023-01204-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10142-023-01204-y