
Abstract
Cytoplasmic male sterility (CMS) has been widely used in crop cross breeding. There has been much research on wheat CMS. However, the correlation between S-type CMS and mitochondrial genome remains elusive. Herein, we sequenced the mitochondrial genome of wheat CMS line and compared it with the maintainer line. The results showed that the mitochondrial genome of CMS line encoded 26 tRNAs, 8 rRNAs, and 35 protein-coding genes, and the cob encoding complex III in which the protein coding gene is mutated. This protein is known to affect reactive oxygen (ROS) production. The analysis of ROS metabolism in developing anthers showed that the deficiency of antioxidants and antioxidant enzymes in the sterile system aggravated membrane lipid oxidation, resulting in ROS accumulation, and influencing the anther development. Herein, cob is considered as a candidate causative gene sequence for CMS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) is one of the most widely distributed and largest food crops in the world. In the face of growing population demand and consumption, it is important to increase global wheat production in a sustainable and effective manner. Heterosis (hybrid vigor) is a natural phenomenon in which the hybrid offspring of genetically diverse individuals show better yield, stress resistance, and fertility than their parents (Fu et al. 2014). To date, heterosis has been applied to rice (Oryza sativa L.), maize (Zea mays L.), rape (Brassica napus L.), soybean (Glycine max (Linn.) Merr.), and other crops; in general, hybrid varieties have higher and more consistent yields than common varieties (Longin et al. 2012; Cerna et al. 1997). Wheat is a self-pollinated plant, and its hybrid breeding requires blocking self-pollination, which can be achieved by genetic male sterility, photoperiothermic male sterility, chemical agents, or cytoplasmic male sterility (Singh et al. 2014).
Cytoplasmic male sterility (CMS) is a widespread phenomenon in higher plants that occurs due to the complex interaction between the nucleus and cytoplasm (Bohra et al. 2016), manifested by maternally inherited, pollen failure, and normal pistils. Sterility genes are encoded by mitochondrial genomes, while most fertility restorer genes are encoded by nuclear genomes (Xiao et al. 2020). Therefore, the application of CMS in hybrid breeding has become a major international trend. CMS has been found in more than 150 plants, such as maize (Zea mays L.), rice, rape, beets (Beta vulgaris L.), carrots (Daucus carota L.), onions (Allium cepa L.), sunflower (Helianthus annuus L.), and wheat (Carlsson et al. 2008).
The mitochondria are semi-autonomous organelles with their own genetic material and systems and are the main place where cells undergo aerobic respiration and are known as the powerhouse of the cell. Unlike chloroplast genome, mitochondrial genome sizes vary widely among eukaryotes, ranging from 6 kb in plasmodium to 200–2000 kb in higher plants (Palmer and Herbon 1987). Plant mitochondrial DNAs (mtDNAs) have remarkable features that distinguish them from other species. Higher plants harbor large mtDNAs that are highly variable, and genomes vary greatly in size and structure even before close relatives (Tang et al. 2015; Chen et al. 2017a). In most plant species, the mtDNA gene sequences evolve very slowly, as compared to animal mtDNA sequences. Plant mtDNAs have a high frequency of homologous recombination, which makes it easy to rearrange mtDNAs; when not lethal, it may result in CMS (Gualberto et al. 2014).
At present, CMS in wheat mainly comprise T (T.timopheevii), P (Primepi), V (Aegilops Ventricosa), K (Aegilops Kotchyi), and S (T.spelta) types of cytoplasms and so on (Wu et al. 2010; Sun et al. 1985). It has been found that abnormal anther development of K-type CMS wheat may be related to sucrose metabolism (Ba et al. 2019). By sequencing the mitochondrial genome of K-type CMS, it was found that the K-type CMS line was missing rps5 gene (Huitao Liu et al. 2011). Proteomic analysis of P-type CMS in wheat suggested that P-type CMS may be a result of cellular dysfunction caused by the disturbance of carbohydrate metabolism, inadequate energy supply, and protein synthesis (Zhang et al. 2021). Proteomics approaches found that V-type CMS in wheat may be correlated to premature programmed cell death (Guo Baojian et al. 2011). Abnormal expression of chalcone synthase (CHS) gene has been found in S-type CMS, and abnormal methylation has also been found in leaves of S-type CMS during seedling stage (Ba et al. 2014, 2017). At present, there is much research on CMS in wheat, but there is few research on the relationship between CMS and mitochondrial genome in wheat. S-type cytoplasmic interaction male sterility in wheat (S stands for sterile cytoplasm of wheat with T. spelta), the fertility transition is relatively easy, the restoration source is wide, and the restorer genes are basically distributed in common wheat species, which can create new restorer lines through accumulation of hybrid and has a good application prospect (Zhang et al. 1986; Ba et al. 2014). To study the mechanism of S-type CMS in a more systematic way, we sequenced and assembled the complete mitochondrial genome of S-type CMS. In addition, we compared and analyzed the complete mitochondrial genome of CMS and its maintainer and clarified their collinearity, InDels, SNPs, and Ka/Ks. The results provide insight into the intricate relationships between mtDNA and CMS generation and may stimulate further research.
Materials and methods
Plant materials
A S-type CMS line (S1376A) and its maintainer line (1376B) were provided by the Wheat Breeding Center, Northwest A&F University. All cultivars were bred successively over 20 years so that the CMS line is stable, and its nuclear background is the same as that of the maintainer. Plants were cultivated in experimental fields at Huaibei Normal University, Huaibei City (longitude 116.80′E, latitude 33.95′N), Anhui Province, China, in November 2020.
Microscopic observation
Fresh anthers were collected after the wheat started heading, and the development stage of microspores was determined by acetic acid magenta dyeing tablet method combined with cytological microscopy. Anther fertility was identified with iodine-potassium iodide (KI-I2), and the collected flowers were stained with a 1% KI-I2 solution and observed under an optical microscope. Healthy and fertile pollen grains are spherical and stained dark, sterile pollen grains atrophy and remain stainless.
Sequencing and assembly of mitochondrial genome
Approximately 5 g of fresh wheat leaves was collected, and mtDNA was extracted by an improved method (Chen et al. 2011). Then, the mitochondrial genome was sequenced and assembled applying second- and third-generation sequencing platforms. The second-generation sequencing platform was Illumina Novaseq 6000 (Genepioneer Biotechnologies), using the fastp (version 0.20.0, https://github.com/OpenGene/fastp) software to the original data filtering; the filter criteria is as follows: The sequencing joints and primer sequences in reads were intercepted; reads with an average mass value less than Q5 were filtered out; filter out reads with N numbers more than 5, then get high-quality reads. The third generation is the nanopore sequencing platform; the software LoRDEC (V0.6) was used to calibrate the third-generation original data with the second-generation data (Salmela and Rivals 2014).
Mitochondrial genome assembly involves several processes. Firstly, using the third-generation assembly software canu to splice the original third-generation data, then get contig sequences (Koren et al. 2017). Secondly, using blast v2.6 (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to align contig sequences in plant mitochondrial gene database (mitochondrial gene sequences of published species on NCBI), the sequence on the contrast pair was stretched and cyclized. Thirdly, using NextPolish1.3.1 (https://github.com/Nextomics/NextPolish) to correct the assembly results with second- and third generation-data, the final assembly results are obtained after a manual inspection and correction (Hu et al. 2020).
Genome annotations and sequence analysis
Mitochondrial genetic structure annotation was divided into the following steps: (1) The encoding protein and rRNA used BLAST to align the published plant mitochondrial sequences as refs; further manual adjustments are made for related species; (2) tRNA was annotated using tRNAscanSE (http://lowelab.ucsc.edu/tRNAscan-SE/) (Chan and Lowe 2019); and (3) OpenReading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to annotate ORF, the shortest length was set to 102 bp, and redundant sequences and sequences with overlap with known genes were excluded. Sequence alignments greater than 300 bp are annotated to the NR library. To obtain more accurate annotation results, the above results were checked and manually corrected. Then, the mitochondrial genome was mapped using OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).
Sequence analysis includes relative synonymous codon usage (RSCU) and scattered in repeated sequence analysis. The RSCU calculation method is (the number of one of the codons encoding an amino acid/the number of all codons encoding the amino acid)/(1/the type of codon encoding the amino acid), that is (the actual frequency of use of the codon/the theoretical frequency of use of the codon). Scattered repeats sequences were identified using the Vmatch v2.3.0 software (http://www.vmatch.de/) combined with Perl scripts. Its parameters were set as minimum length = 30 bp, amming distance = 3, and there were four recognition forms: forward, palindromic, reverse, and complement.
Comparative analysis of S1376A and 1376B
Firstly, the mitochondrial genomes of S1376A and 1376B were globally compared, and then the SNPs and InDels were found by using the AUTO pattern alignment of the MAFFT V7.427 software. KaKs Calculator 2.0 was used to perform Ka/Ks calculation for maFFT-aligned sequences. Using the Mauve software, the mitochondrial genome of S1376A was compared with that of its maintainer line (Rissman et al. 2009). Using DNASP5 for PI calculation, set the parameter as window 400 bp and step size 200 bp. Signal peptides were predicted using signalP-4.1 EUK mode.
Determination of O2.−, H2O2, and MDA content
The anthers of S1376A and 1376B were collected at the uninucleate stage, binucleate stage, and trinucleate stage and prepared in the refrigerator at − 80 °C. The content of superoxide radical (O2−) was determined by hydroxylamine oxidation method (Lukatkin 2002). Determination of hydrogen peroxide (H2O2) was by titanium chloride method (Liu et al. 2010). The determination of malondialdehyde (MDA) content was based on the method of Hodges et al. and improved (Hodges 1999).
Determination of AsA and GSH content
The content of ascorbic acid (AsA) was determined by the Kampfenkel method, and glutathione (GSH) was determined by the DTNB cyclic method (Nagalakshmi and Prasad 2001; Kampfenkel et al. 1995).
Enzyme activity determinations
The activity of superoxide dismutase (SOD) were determined by the nitrate blue tetrazole (NBT) photoreduction method and the guaiacol method (Shen et al. 2014). The peroxidase (POD) activity was determined by guaiacol colorimetry (Hui Fang et al. 2012). Catalase (CAT) activity was mainly determined by the UV absorption method of Chance et al. (1955); ascorbate peroxidase (APX) activity was mainly determined by the UV absorption method (Miyake et al. 2006). Glutathione reductase (GR) activity was determined based on the modified method of Alonso GOnzález and Lynch 1998). Monodehydroascorbate reductase (MDHAR) activity was determined based on the converted method of Knörzer et al. (2008).
Results
Cytological observation of S1376A
To determine anther fertility, KI-I2 was used to stain the anther. We found that the pollen grains of 1376B were rich in starch (Fig. 1e), while the pollen grains of sterile line had no starch (Fig. 1a), indicating that sterile line S1376A was completely aborted. To determine the anther stage, we used acetic acid magenta staining to find that the anther was in uninucleate stage when the ear was just drawn out but not completely drawn out (Fig. 1b, f), in binucleate stage when the head sprouting completely (Fig. 1c, g), and in trinucleate stage after a period when the head sprouting completely (Fig. 1d, h).

Pollen development period and wheat fertility. Pollen grains of sterile and maintainer lines are stained with I2-KI solution (a, e). The anther structures of b and f; c and g; d and h were mononuclear, binucleate stage and trinucleate stage, respectively
Characteristics of mitochondrial genome assembly in S1376A
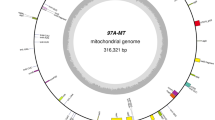
To investigate the specificity of S1376A mitochondrial genome, we sequenced it using the Llumina Novaseq 6000 and the nanopore sequencing platform. The mitochondrial genome of S1376A was assembled as a single molecule with a length of 452,638 bp, the content of GC was 44.36%, AT skew is 0.001, and GC skew is 0.002 (Fig. 2). The percentages of clean data bases with mass values greater than or equal to 20 and 30 were 98.05% and 94.18%, respectively. The mitochondrial genes of the CMS line were annotated with BLAST and tRNAscanSE, and 69 genes were identified (Table 1), including 26 tRNA genes, 8 rRNA genes (rrn5 (3), 18 (3), 26 (2)), and 35 protein-coding genes. There are several functions of protein-coding genes; one gene for maturase (matR), one gene for transport membrance protein (mttB), nine genes for complex I (nad1-7, 9, 4L), one gene for complex III (cob), three genes of complex IV (cox1-3), seven genes for complex V (atp1, 4, 6 (2), 8 (2), 9), four genes for cytohrome c biogenesis (ccmB, ccmC, ccmFN, ccmFc), and nine genes for ribosomal proteins (rpl5, 16, rps1, 2, 4, 7, 12–14). Of these, rrn26, rps3, and rps19 are pseudogenes; these three pseudogenes may be non-functional residues formed during the transformation of gene families, which are DNA fragments that have lost their gene functions and cannot be transcribed into normal mRNA and thus cannot be expressed. Through annotation of ORF, 2634 ORFs are found to be CDS, among which 1327 are forward encoding and 1307 are reverse encoding. There are 82 ORFs of length greater than 300 bp; through blast, it revealed that there were 7 with high homology (score > 400). In particular, orf1637 was highly similar fatty acyl-CoA reductase 2-like in Aegilops tauschii Coss., and orf499 was also highly similar ribosomal protein S7 in maize chloroplasts. In S1376A, there were 84 unique ORFs, 42 were forward and 42 were backward. Only orf1079, orf2232, orf2650, orf342, and orf958 were larger than 300 bp; however, annotation and blast showed that they did not encode protein.

Mitochondrial genome map. Genes that encode forward are located on the outside of the circle, and those that encode backward are located on the inside. The gray circle inside represents the GC content
Relative synonymous codon usage (RSCU) analysis of PCGs
Because of the degeneracy of codons, each amino acid corresponds to at least one codon and at most six codons. The codon utilization rate of different species and different organisms diverges greatly. This unequal use of synonymous codons is called relative synonymous codon usage. It is believed to be the result of natural selection, mutation, and genetic drift. We used our own Perl script to filter unique CDS and calculate the RSCU value (Fig. 3 and Supplementary Fig. S1). Our analysis of S-type CMS mitochondrial genome showed that there were 31 codons with RSCU > 1 (Supplementary Table S1), indicating that these codons were preferentially used in the genome. The AGA bias of the codon encoding aspartic acid was the highest with RSCU of 1.55. The second is the isoleucine codon CAU, and the RSCU value is 1.54. Among the preferentially used codons, all codons ended with U (16 of 31) or A (12 of 31); two of the other three codons end in C and one ends in G.

RSCU pie chart. The height of the outermost cylinder is RSCU, the inner layer is the amino acid, and the innermost three layers are codons
Repeated sequence analysis
There were 9 large repeating sequences in the mitochondrial genome of CMS line, that is, the sequences with length ≥ 500 bp and the similarity more than 90%. The longest sequence length was 9882 bp, and the similarity was 99.98%. The shortest sequence length was 1362 bp, and the similarity was 90.05%. The similarity of four repeat sequences reached 100%, with lengths of 7035 bp, 5469 bp, 2045 bp, and 1634 bp (Supplementary Table S2).
Scattered repeats sequence, which are different from tandem repeats, are distributed in a dispersed manner in the genome; such DNA sequences are generally moderately repetitive. We used the vmatch V2.3.0 software to identified repeated sequences, and the identification forms were divided into forward, palindromic, reverse, and complement. According to the analysis results of scattered repeats sequence, it was found that there were only two forms of scattered repeats sequence in the mitochondrial genome of sterile wheat, namely forward repeats and reverse repeats. Only a few of the two types of repeat lengths were above 40 bp, and the repeats between 80–89 bp (F:2, P: 5) and 90–99 bp (F:1, P: 4) were the least. The longest scattered repeats sequence was forward repeat, and the length was 7035 bp, which encoded two rrn26 genes. Most of the other scattered repeat sequences were between 30 and 39 bp (F: 99, P: 95) (Fig. 4, Supplementary Table S3). The results showed that there were 20 forward repeats and 29 palindromic repeats in the scattered repeats sequence of 30 bp. Eight genes were encoded in the 49 scattered repeats sequence, including two nad7, two nad4 (intron), two rps3 (intron), one cox1, one ccmC, and 46 intergenic spacers (IGS) (Supplementary Table S4).

Graph of the distribution and length of the scattered repeats. The distribution of four kinds of scattered repeats on the genome (A). The two black lines represent the genome, and the same repeats are connected by line segments. The abscissa of B is the type of scattered repeats, and the ordinate is the number of scattered repeats. F, forward repetition; P, palindromic repetition; R, reverse repetition; C, complementary repetition
Collinearity analysis of S1376A and 1376B
Collinearity is mainly used to describe the location of genes on the same chromosome, that is, the type of genes and the conservation of relative order among distinctive species differentiated from the same ancestral type (that is, the homology of genes and the sequence of genes). We used the mauve software to analyze the mitochondrial genomes of S1376A and 1376B, and the results showed that neither sequence inversion nor rearrangement occurred in both genomes, only slight differences in gene location and type (Fig. 5). This indicated that the sequences of these two kinds of wheat had good collinearity and high homology. Specific differences in the gene type and location require further analysis.

Collinearity of the maintainer line and the CMS line
Identification of InDels and SNPs in S1376A and 1376B
Insertions/deletions (InDels) refer to the insertion or deletion of nucleotide fragments of different sizes at the same site of the genome between related species or individuals of the same species. Single nucleotide polymorphisms (SNPs) are the polymorphism of nucleic acid sequence caused by changes in a single nucleotide base, including substitution, transposition, deletion, and insertion. After we searched for SNPs and InDels in the S-type CMS line and the maintainer line, we identified 46 InDels and 303 SNPs (Supplementary Table S5). These InDels distributed in 39 orfs (Table 2) and 3 PCGs (cob, nad1, nad2(intron)). There were just 13 SNP-encoding genes: two SNPs for cox3, one SNP for matR, one SNP for nad4(intron), one SNP for rps4, one SNP for trnF, two SNPs for trnS, two SNPs for nad7(intron), and three SNPs for trnW (Table 3). Studies have shown that Indian cauliflowers’ (Brassica oleracea L. var. botrytis L.) cytoplasmic genetic variation is related to SNPs and InDels (Singh et al., 2021). We think that the genetic variation of the S-type CMS line may also be related to 303 SNPs and 46 InDels.
Subsequently, Pi analysis calculated the nucleic acid diversity of each gene. We used DNASP5 for PI calculation and set the parameter as window 400 bp and step size 200 bp. The calculation results revealed that the nucleotide diversity value was 0.00067, and the number of polymorphic sites was 302 (Supplementary Table S6). Among them, 211 fragments had nucleotide diversity; the maximum was 0.0725, occupying 29 polymorphic sites, and the minimum was 0.0025, occupying 134 polymorphic sites (Fig. 6).

Nucleic acid diversity
Ka/Ks analysis of S1376A and 1376B
We calculated the Ka/Ks of protein-coding genes and found that non-synonymous mutations occurred in cob, rps4, and cox3, with mutation rates of 0.0016, 0.0011, and 0.0032, respectively. The synonymous mutation rates of cob and matR were 0.0065 and 0.0025, respectively. Only cob had non-synonymous mutation and synonymous mutation, and its Ka/Ks value was 0.24 < 1 (Table 4). Ka/Ks represents the ratio between non-synonymous substitutions (Ka) and synonymous substitutions (Ks). This ratio determines whether there is selection pressure on the protein-coding gene. Ka/Ks > 1 indicates a positive selection, Ka/Ks < 1 indicates a negative selection, and Ka/Ks ≈ 1 indicates a neutral evolution (Li et al. 2009). This showed that cob was negatively selected during evolution. Whether negative selection of cob can induce pollen abortion needs to be verified in the S-type CMS line and the maintainer line.
Analysis of physiological indicators
Ubichinol cytochrome c reductase encoded by cob is complex III in the mitochondrial respiratory chain. It has been convinced that complex III is the main source of ROS (Mazat et al. 2020; Brand 2016). Herein, we measured the relevant indicators, and the results showed that the content of H2O2 in S1376A and 1376B was significantly different in uninucleate stage and binucleate stage, and the content of H2O2 in S1376A in binucleate stage was more advanced than that in 1376B. The content of O2− in S1376A and 1376B was significantly different only at binucleate stage, and the content of O2− was higher in S1376A at each stage. The content of MDA in S1376A and 1376B was significantly different in all three stages, and the accumulation of H2O2 in S1376A was more. SOD activity in anthers increased in 1376B and decreased in S1376A. The SOD activity in 1376B was significantly higher than that in S1376A at the binucleate and trinucleate stages. The POD activity in anthers was significantly different among different cultivars at the same stage and increased in S1376A and then decreased in 1376B. There were significant differences in CAT activity between S1376A and 1376B anthers at binucleate stage, and CAT activity in 1376B was significantly higher than that in S1376A at binucleate stage (Fig. 7). The contents of ASA and GSH in 1376B were significantly higher than these in S1376A at binucleate stage. The APX activity maintained significantly higher at binucleate stage than in S1376A, and the difference was significant at other stages. GR activity in 1376B was prominently higher than that in S1376A at uninucleate stage and binucleate stage, while MDHAR activity was notably higher in 1376B than that in the S1376A only at binucleate stage (Fig. 8). In summary, the contents of antioxidant substances in S1376A were deficient, and the activity of antioxidant enzymes was low, especially at the binucleate stage.

ROS metabolism in the anthers of maintainer and sterile lines at different periods. Different lowercase letters indicate that different varieties have significant differences at the level of 0.05, the same as below

Effects of ascorbate–glutathione cycle on the anthers of maintainers and sterile lines at different stages
Discussion
The mitochondria are important energy supply sites for maintaining normal functions of cells. In addition, the mitochondria also participate in cell differentiation, apoptosis, cell information transmission, and other processes. The mitochondria themselves carry genetic material that coordinates with the nucleus to ensure the assembly and function of the large number of proteins in the mitochondrial respiratory chain (Osellame et al. 2012). It has highly variable intergenic regions containing distinct repeats, frequent structural rearrangements, large amounts of gene loss, and a highly variable RNA editing process (Gualberto et al. 2014). CMS is a complex trait which is believed to be related to mitochondrial genome and may be influenced by the evolution pattern of mitochondrial genome and gene transfer between organelles and nuclear ventricles in plant cells (Chen et al. 2017b). In previous research on wheat CMS, there have been a lot of research on male sterility, most of which focused on proteomics (Geng et al. 2018), cytology (Martin et al. 2010), and epigenetics (Ba et al. 2014), but there is little research on mitochondrial genome sequencing. At present, there are only three kinds of CMS type wheat mitochondrial genome sequencing. One was K-type CMS, which showed that rpl5 was absent in Ks3 and trnH was absent in Km3, twenty-two unique orfs were predicted in Ks3, representing potential candidate genes for K-type CMS (Huitao Liu et al. 2011). One was Al-type CMS, and the results showed that 12 orfs were selected as candidate causative gene sequences, among which orf279 was considered as the candidate causative gene sequence of Al-type CMS line (Hao et al. 2021). One was T-type CMS, the mechanism of this type of CMS has been determined to be due to the presence of orf279 (Melonek et al. 2021). In this research, S-type CMS was selected as the research material, and its mitochondrial genome sequence was compared and analyzed with that of the maintainer line, which provided a reference for further exploring the mechanism of S-type CMS.
Mitochondrial genome characteristics of CMS line
Unlike animal mitochondrial genome and plant chloroplast genome, their structure is conservative, and the gene arrangement is compact and very stable. Plant mitochondrial genomes vary greatly in size and structure and are sparsely arranged (Li et al. 2020a; Alverson et al. 2010). Repeated sequences are common in plant mitochondrial genomes, and most of the plant mitochondrial genomes are large, probably because there are many repeat sequences (Lowe and Chan 2016; Li et al. 2020a). However, there are different situations, Cucurbita mitochondrial genome 38% of the genome comprises repeat sequences, but its full mtDNA length is 371 kb (Alverson et al. 2010). The length of Vitis mtDNA reached about 773 kb, but only 7% of the genome that comprises repeat sequences were found (Goremykin et al. 2009). Our sequencing results showed that the length of S-type CMS mitochondrial genome was 452,638 bp, encoding 69 genes in total. The length of long repeats was 40,400 bp, and the length of scattered repeats was 60,563 bp, accounting for 8.9% and 13.4% of the mtDNA, respectively. The total length of repeating sequences was much lower than that of K-type CMS (Huitao Liu et al. 2011).
Comparison of mtDNA between the CMS line and the maintainer line
The mitochondrial genome length of S-type CMS in wheat was larger than that of maintainer, which was found in the mitochondrial genome of Nicotiana tabacum and Brassica rapa (Wang et al. 2020; Li et al. 2020b). No sequence inversion or rearrangement occurred in the two genomes. The sequences of CMS line and maintainer line were collinearity and homology were high. However, 46 InDels (39 orfs and 3 genes) and 303 SNPs (13 genes) were found by CMS and maintainer mtDNA comparative analysis. A similar phenomenon was found in soybeans. The mtDNA study of soybean CMS line found 209 SNPs and 110 InDels (He et al. 2021). We believe that SNPs and InDels may affect the fertility of wheat. The same study of Indian cauliflower (Brassica oleracea L. var. botrytis L.) mtDNAs found that its cytoplasmic genetic alteration was related to SNPs and InDels (Singh et al. 2021). Our Ka/Ks analysis showed that only the cob gene has both synonymous mutations and non-synonymous mutations. At present, some studies have shown that cob is related to CMS phenomenon. An unusual transcription pattern of mitochondrial cob gene was found in onion (Allium cepa L.) with S-type CMS, sequencing of mitochondrial cob gene revealed that chloroplast DNA sequence was inserted into the upstream region of cob in S-type cytoplasm (Sato 1998). In the cytoplasm of “Pampa” in rye (Secale cereale L.), which contains an additional cob-homologous transcript that might causally be correlated to the CMS phenotype (Dohmen et al. 1994). In our research, negative selection occurred in the mitochondrial cob gene of S-type male sterile line of wheat. After comparing the sequence of this gene, we discovered that several bases were inserted in a sequence of this gene, resulting in code shift and backward movement of the stop codon. We speculate that this might causally be correlated to the S-type CMS phenotype in wheat.
Cob is the differential gene between S1376A and 1376B, and the ubichinol cytochrome c reductase encoded by cob is complex III in the mitochondrial respiratory chain, and its function in the mitochondrial respiratory chain is to oxidize a ubiquinone in its diminished state and transfers one pair of electrons to cytochrome c. The four electrons of cytochrome c are then transferred to oxygen by complex IV (cytochrome c oxidase) to form H2O (Zhao et al. 2019; Ji et al. 2013). Studies have shown that complex III is the main source of ROS, mainly in the formation of H2O2 and O2− (Mazat et al. 2020; Brand 2016). MDA reflects the main index of membrane lipid peroxidation. In the binucleate stage, we found that the contents of H2O2 and MDA in the S-type CMS line were significantly higher than those in 1376B, indicating that ROS was accumulated in S1376A. ROS are toxic by-products produced during aerobic metabolism, which may lead to cell death if not clear in time (Maxwell et al. 2002). There are many mechanisms of ROS elimination in organisms. One mechanism involves antioxidants, such as GSH and AsA. Another mechanism relies on the protective system of antioxidant enzymes, including SOD, POD, CAT, and APX (Yang et al. 2018). GR and MDHAR engage in ascorbate–glutathione cycle and play a vital role in scavenging the accumulation of ROS (Miller et al. 2010). The research found that the content of antioxidant substances in the anthers of S1376A was deficient, and the activity of antioxidant enzymes was low, leading to the accumulation of ROS and MDA. Similar results were found in other plants. The activities of flavonoids and antioxidant enzymes in flower buds of soybean CMS lines were lower than those of maintainers (Ding et al. 2019). ROS levels were higher in the rice CMS line than in the maintainer line (Yan et al. 2014). More ROS were accumulated in the anthers of cotton CMS lines (Jiang et al. 2007). In pepper (Capsicum annuum L.) CMS line, the activity of antioxidant enzyme was lower than that of maintainer, and ROS accumulated more (Deng et al. 2012). These results indicate that ROS metabolism may be related to male sterility, especially in binucleate stage. However, whether this phenomenon is related to cob mutation still needs to be further determined.
References
Alonso Gonza´lez KLS, Lynch JP (1998) Light and excess manganese: implications for oxidative stress in common bean. Plant Physiol 118:493–503. https://doi.org/10.1104/pp.118.2.493
Alverson AJ, Wei X, Rice DW, Stern DB, Barry K, Palmer JD (2010) Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol 27(6):1436–1448. https://doi.org/10.1093/molbev/msq029
Ba Q, Zhang G, Li G, Zhou L, Zhang G, Fu Z, Chen C, Song Y (2017) Analysis of chalcone synthase gene in chemically induced male sterility (CIMS) in wheat (Triticum aestivum L). Indian J Genet Plant Breed 77:508. https://doi.org/10.5958/0975-6906.2017.00067.0
Ba Q, Zhang G, Niu N, Ma S, Wang J (2014) Cytoplasmic effects on DNA methylation between male sterile lines and the maintainer in wheat (Triticum aestivum L). Gene 549(1):192–197. https://doi.org/10.1016/j.gene.2014.01.001
Ba Q, Zhang L, Li G, Zhang G, Liu H, Fu Z (2019) Altered sucrose metabolism and gene regulation in wheat (Triticum aestivum) K-type cytoplasmic male sterility. Crop Pasture Sci 70(3):204–210. https://doi.org/10.1071/CP18517
Bohra A, Jha UC, Adhimoolam P, Bisht D, Singh NP (2016) Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep 35(5):967–993. https://doi.org/10.1007/s00299-016-1949-3
Brand MD (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100:14–31. https://doi.org/10.1016/j.freeradbiomed.2016.04.001
Carlsson J, Leino M, Sohlberg J, Sundström JF, Glimelius K (2008) Mitochondrial regulation of flower development. Mitochondrion 8(1):74–86. https://doi.org/10.1016/j.mito.2007.09.006
Cerna FJ, Cianzio SR, Rafalski A, Tingey S, Dyer D (1997) Relationship between seed yield heterosis and molecular marker heterozygosity in soybean. Theor Appl Genet 95(3):460–467. https://doi.org/10.1007/s001220050583
Chan PP, Lowe TM (2019) tRNAscan-SE: searching for tRNA genes in genomic sequences. In: Kollmar M (ed) Gene prediction: methods and protocols, vol 1962. Springer New York, New York, NY, pp 1–14. https://doi.org/10.1007/978-1-4939-9173-0_1
Chance B, Maehly AC (1955) The assay of catalases and peroxidases. In: Methods in enzymology, vol 2. Academic Press, pp 764–775. https://doi.org/10.1016/S0076-6879(55)02300-8
Chen J, Guan R, Chang S, Du T, Zhang H, Xing H (2011) Substoichiometrically different mitotypes coexist in mitochondrial genomes of Brassica napus L. PLoS One 6(3):e17662. https://doi.org/10.1371/journal.pone.0017662
Chen Z, Nie H, Grover CE, Wang Y, Li P, Wang M, Pei H, Zhao Y, Li S, Wendel JF, Hua J (2017) Entire nucleotide sequences of Gossypium raimondii and G arboreum mitochondrial genomes revealed A-genome species as cytoplasmic donor of the allotetraploid species. Plant Bio 19(3):484–493. https://doi.org/10.1111/plb.12536
Chen Z, Zhao N, Li S, Grover CE, Nie H, Wendel JF, Hua J (2017) Plant mitochondrial genome evolution and cytoplasmic male sterility. Crit Rev Plant Sci 36(1):55–69. https://doi.org/10.1080/07352689.2017.1327762
Hodges DM, Delong JM, Forney CF, Prange RK (1999) Improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds. Planta 207:604–611. https://doi.org/10.1007/s004250050524
Deng M-H, Wen J-F, Huo J-L, Zhu H-S, Dai X-Z, Zhang Z-Q, Zhou H, Zou X-X (2012) Relationship of metabolism of reactive oxygen species with cytoplasmic male sterility in pepper (Capsicum annuum L.). Sci Hortic 134:232–236. https://doi.org/10.1016/j.scienta.2011.10.027
Ding X, Wang X, Li Q, Yu L, Song Q, Gai J, Yang S (2019) Metabolomics studies on cytoplasmic male sterility during flower bud development in soybean. Int J Mol Sci 20(12):2869. https://doi.org/10.3390/ijms20122869
Dohmen G, Hessberg H, Geiger HH, Tudzynski R (1994) CMS in rye’ comparative RFLP and transcript analyses of mitochondria from fertile and male-sterile plants. Theor Appl Genet 89:1014–1018. https://doi.org/10.1007/BF00224532
Fu D, Xiao M, Hayward A, Fu Y, Liu G, Jiang G, Zhang H (2014) Utilization of crop heterosis: a review. Euphytica 197(2):161–173. https://doi.org/10.1007/s10681-014-1103-7
Geng X, Ye J, Yang X, Li S, Zhang L, Song X (2018) Identification of proteins involved in carbohydrate metabolism and energy metabolism pathways and their regulation of cytoplasmic male sterility in wheat. Int J Mol Sci 19 (2). https://doi.org/10.3390/ijms19020324
Goremykin VV, Salamini F, Velasco R, Viola R (2009) Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol Biol Evol 26(1):99–110. https://doi.org/10.1093/molbev/msn226
Gualberto JM, Mileshina D, Wallet C, Niazi AK, Weber-Lotfi F, Dietrich A (2014) The plant mitochondrial genome: dynamics and maintenance. Biochimie 100:107–120. https://doi.org/10.1016/j.biochi.2013.09.016
Guo Baojian, Li Peng, Mou Qiuhuan (2011) Differential mitochondrial proteome expression between v-cms of wheat and hybrid F_1. Mol Plant Breed 9(3):302–308. CNKI:SUN:FZZW.0.2011–03–010.
Hao M, Yang W, Lu W, Sun L, Shoaib M, Sun J, Liu D, Li X, Zhang A (2021) Characterization of the mitochondrial genome of a wheat AL-type male sterility line and the candidate CMS gene. Int J Mol Sci 22(12):6388. https://doi.org/10.3390/ijms22126388
He T, Ding X, Zhang H, Li Y, Chen L, Wang T, Yang L, Nie Z, Song Q, Gai J, Yang S (2021) Comparative analysis of mitochondrial genomes of soybean cytoplasmic male-sterile lines and their maintainer lines. Funct Integr Genomics 21(1):43–57. https://doi.org/10.1007/s10142-020-00760-x
Hu J, Fan J, Sun Z, Liu S (2020) NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36(7):2253–2255. https://doi.org/10.1093/bioinformatics/btz891
Hui Fang QZ, He Y, Li X-l (2012) Detection of activity of POD in tomato leaves based on hyperspectral imaging technology. Spectrosc Spectr Anal 32(8):2228–2233. https://doi.org/10.3964/j.issn.1000-0593(2012)08-2228-06
Huitao L, Cui P, Kehui Z, Qiang L, Zhuo G, Guo X, Ding F, Yang W, Liu D, Hu S, Yu J, Aimin Z (2011) Comparative analysis of mitochondrial genomes between a wheat K-type cytoplasmic male sterility (CMS) line and its maintainer line. BMC Genomics 12(1):1–14. https://doi.org/10.1186/1471-2164-12-163
Ji J, Huang W, Yin C, Gong Z (2013) Mitochondrial cytochrome c oxidase and F1Fo-ATPase dysfunction in peppers (Capsicum annuum L) with cytoplasmic male sterility and its association with orf507 and Psiatp6–2 genes. Int J Mol Sci 14 (1):1050–1068. https://doi.org/10.3390/ijms14011050
Jiang P, Zhang X, Zhu Y, Zhu W, Xie H, Wang X (2007) Metabolism of reactive oxygen species in cotton cytoplasmic male sterility and its restoration. Plant Cell Rep 26(9):1627–1634. https://doi.org/10.1007/s00299-007-0351-6
Kampfenkel K, Vanmontagu M, Inze D (1995) Extraction and determination of ascorbate and dehydroascorbate from plant tissue. Anal Biochem 225(1):165–167. https://doi.org/10.1006/abio.1995.1127
Knörzer O, Burner J, Boger P (2008) Alterations in the antioxidative system of suspension-cultured soybean cells (Glycine max) induced by oxidative stress. Physiol Plant 97:388–396. https://doi.org/10.1034/j.1399-3054.1996.970225.x
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27(5):722–736. https://doi.org/10.1101/gr.215087.116
Li J, Zhang Z, Vang S, Yu J, Wong GK, Wang J (2009) Correlation between Ka/Ks and Ks is related to substitution model and evolutionary lineage. J Mol Evol 68(4):414–423. https://doi.org/10.1007/s00239-009-9222-9
Li M, Chen L, Tang D, Liao X, Kong X, Li B, You J, Zhou R (2020a) Discovery of four novel ORFs responsible for cytoplasmic male sterility (CMS) in cotton (Gossypium hirsutum L.) through comparative analysis of the mitochondrial genomes of four isoplasmic lines. Agronomy 10 (6). https://doi.org/10.3390/agronomy10060765
Li P, Zhang D, Su T, Wang W, Yu Y, Zhao X, Li Z, Yu S, Zhang F (2020b) Genome-wide analysis of mRNA and lncRNA expression and mitochondrial genome sequencing provide insights into the mechanisms underlying a novel cytoplasmic male sterility system, BVRC-CMS96. Brassicarapa Theor Appl Genet 133(7):2157–2170. https://doi.org/10.1007/s00122-020-03587-z
Liu X, Williams CE, Nemacheck JA, Wang H, Subramanyam S, Zheng C, Chen MS (2010) Reactive oxygen species are involved in plant defense against a gall midge. Plant Physiol 152(2):985–999. https://doi.org/10.1104/pp.109.150656
Longin CF, Muhleisen J, Maurer HP, Zhang H, Gowda M, Reif JC (2012) Hybrid breeding in autogamous cereals. Theor Appl Genet 125(6):1087–1096. https://doi.org/10.1007/s00122-012-1967-7
Lowe TM, Chan PP (2016) tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44(W1):W54-57. https://doi.org/10.1093/nar/gkw413
Lukatkin AS (2002) Contribution of oxidative stress to the development of cold-induced damage to leaves of chilling-sensitive plants: 1. Reactive oxygen species formation during plant chilling. Russian J Plant Physiol 49(5):622–627. https://doi.org/10.1023/a:1020232700648
Martin AC, Atienza SG, Ramirez MC, Barro F, Martin A (2010) Molecular and cytological characterization of an extra acrocentric chromosome that restores male fertility of wheat in the msH1 CMS system. Theor Appl Genet 121(6):1093–1101. https://doi.org/10.1007/s00122-010-1374-x
Maxwell DP, Nickels R, McIntosh L (2002) Evidence of mitochondrial involvement in the transduction of signals required for the induction of genes associated with pathogen attack and senescence. Plant J 29(3):269–279. https://doi.org/10.1046/j.1365-313X.2002.01216.x
Mazat JP, Devin A, Ransac S (2020) Modelling mitochondrial ROS production by the respiratory chain. Cell Mol Life Sci 77(3):455–465. https://doi.org/10.1007/s00018-019-03381-1
Melonek J, Duarte J, Martin J, Beuf L, Murigneux A, Varenne P, Comadran J, Specel S, Levadoux S, Bernath-Levin K, Torney F, Pichon J-P, Perez P, Small I (2021) The genetic basis of cytoplasmic male sterility and fertility restoration in wheat. Nat Commun 12(1):1036. https://doi.org/10.1038/s41467-021-21225-0
Miller GAD, Suzuki N, Ciftci-Yilmaz S, Mittler RON (2010) Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant, Cell Environ 33(4):453–467. https://doi.org/10.1111/j.1365-3040.2009.02041.x
Miyake C, Shinzaki Y, Nishioka M, Horiguchi S, Tomizawa K-I (2006) Photoinactivation of ascorbate peroxidase in isolated tobacco chloroplasts: Galdieria partita APX maintains the electron flux through the water–water cycle in transplastomic tobacco plants. Plant Cell Physiol 47(2):200–210. https://doi.org/10.1093/pcp/pci235
Nagalakshmi N, Prasad MNV (2001) Responses of glutathione cycle enzymes and glutathione metabolism to copper stress in Scenedesmus bijugatus. Plant Sci 160(2):291–299. https://doi.org/10.1016/S0168-9452(00)00392-7
Osellame LD, Blacker TS, Duchen MR (2012) Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab 26(6):711–723. https://doi.org/10.1016/j.beem.2012.05.003
Palmer JD, Herbon LA (1987) Unicircular structure of the Brassica hirta mitochondrial genome. Curt Genet 11:565–570. https://doi.org/10.1007/BF00384620
Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT (2009) Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25(16):2071–2073. https://doi.org/10.1093/bioinformatics/btp356
Salmela L, Rivals E (2014) LoRDEC: accurate and efficient long read error correction. Bioinformatics 30(24):3506–3514. https://doi.org/10.1093/bioinformatics/btu538
Sato Y (1998) PCR amplification of CMS-specific mitochondrial nucleotide sequences to identify cytoplasmic genotypes of onion (Allium cepa L.). Theor Appl Genet 96:367–370. https://doi.org/10.1007/s001220050750
Shen X, Xiao X, Dong Z, Chen Y (2014) Silicon effects on antioxidative enzymes and lipid peroxidation in leaves and roots of peanut under aluminum stress. Acta Physiol Plant 36(11):3063–3069. https://doi.org/10.1007/s11738-014-1676-8
Singh S, Bhatia R, Kumar R, Behera TK, Kumari K, Pramanik A, Ghemeray H, Sharma K, Bhattacharya RC, Dey SS (2021) Elucidating mitochondrial DNA markers of ogura-based CMS lines in Indian cauliflowers (Brassica oleracea var. botrytis L.) and their floral abnormalities due to diversity in cytonuclear interactions. Front Plant Sci 12:631489. https://doi.org/10.3389/fpls.2021.631489
Singh SP, Srivastava R, Kumar J (2014) Male sterility systems in wheat and opportunities for hybrid wheat development. Acta Physiol Plant 37(1):1713. https://doi.org/10.1007/s11738-014-1713-7
Sun L, Gao Q, Zhang Y, Su Y, Zhou T (1985) Primepi type cytoplasmic male sterility (P-type) research. J Beijing Agri Univ (04):(in Chinese)
Tang M, Chen Z, Grover CE, Wang Y, Li S, Liu G, Ma Z, Wendel JF, Hua J (2015) Rapid evolutionary divergence of Gossypium barbadense and G. hirsutum mitochondrial genomes. BMC Genomics 16:770. https://doi.org/10.1186/s12864-015-1988-0
Wang R, Cai X, Hu S, Li Y, Fan Y, Tan S, Liu Q, Zhou W (2020) Comparative analysis of the mitochondrial genomes of Nicotiana tabacum: hints toward the key factors closely related to the cytoplasmic male sterility mechanism. Front Genet 11:257. https://doi.org/10.3389/fgene.2020.00257
Wu S-W, Gao Q-R, Sun Z, Wang M-T, Sun Z-J, Yuan K, Yu S (2010) Effects of ADP-glucose pyrophosphorylase activity on sterility and development of grain in K, V, and T-cytoplasmic male. Acta Agron Sin 36(6):995–1002. https://doi.org/10.3724/sp.J.1006.2010.00995
Xiao S, Zang J, Pei Y, Liu J, Liu J, Song W, Shi Z, Su A, Zhao J, Chen H (2020) Activation of mitochondrial orf355 gene expression by a nuclear-encoded DREB transcription factor causes cytoplasmic male sterility in maize. Mol Plant 13(9):1270–1283. https://doi.org/10.1016/j.molp.2020.07.002
Yan J, Tian H, Wang S, Shao J, Zheng Y, Zhang H, Guo L, Ding Y (2014) Pollen developmental defects in ZD-CMS rice line explored by cytological, molecular and proteomic approaches. J Proteomics 108:110–123. https://doi.org/10.1016/j.jprot.2014.05.014
Yang L, Wu Y, Zhang M, Zhang J, Stewart JM, Xing C, Wu J, Jin S (2018) Transcriptome, cytological and biochemical analysis of cytoplasmic male sterility and maintainer line in CMS-D8 cotton. Plant Mol Biol 97(6):537–551. https://doi.org/10.1007/s11103-018-0757-2
Zhang G, Yang T, Wang M (1986) Preliminary report on male sterility line of S-type wheat. Shanxi Agri Sci 5:4–7 (In Chinese)
Zhang Y, Song Q, Zhang L, Li Z, Wang C, Zhang G (2021) Comparative proteomic analysis of developmental changes in P-type cytoplasmic male sterile and maintainer anthers in wheat. Int J Mol Sci 22 (4). https://doi.org/10.3390/ijms22042012
Zhao RZ, Jiang S, Zhang L, Yu ZB (2019) Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med 44(1):3–15. https://doi.org/10.3892/ijmm.2019.4188
Acknowledgements
The authors are grateful to all lab members for their useful suggestions, support, and encouragement.
Funding
This work was supported by the Natural Science Foundation of Anhui Province (1708085QC62); Excellent Talents Support Program of Colleges and Universities in Anhui Province (gxyq2020014); Scientific Research Foundation of the Higher Education Institutions of Anhui Province, China (KJ2019A585, KJ2019A0961); Anhui University Collaborative Innovation Project (GXXT-2019–033); and the Doctoral Scientific Research Foundation of Huaibei Normal University (15601047).
Author information
Authors and Affiliations
Contributions
Resources: RW and LZ. Methodology: RW and WW. Formal analysis: QB and RW. Writing—original draft preparation: RW and QB. Writing—review and editing: QB, PZ, and GL. Supervision: QB. Funding acquisition: QB. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Wang, R., Ba, Q., Zhang, L. et al. Comparative analysis of mitochondrial genomes provides insights into the mechanisms underlying an S-type cytoplasmic male sterility (CMS) system in wheat (Triticum aestivum L.). Funct Integr Genomics 22, 951–964 (2022). https://doi.org/10.1007/s10142-022-00871-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-022-00871-7