
Abstract
This study presents the first draft genome of Siganus fuscescens, and thereby establishes the first whole-genome sequence for a species in the Siganidae family. Leveraging both long and short read sequencing technologies, i.e., Oxford Nanopore and Illumina sequencing, we successfully assembled a mitogenome spanning 16.494 Kb and a first haploid genome encompassing 498 Mb. The assembled genome accounted for a 99.6% of the estimated genome size and was organized into 164 contigs with an N50 of 7.2 Mb. This genome assembly showed a GC content of 42.9% and a high Benchmarking Universal Single-Copy Orthologue (BUSCO) completeness score of 99.5% using actinopterygii_odb10 lineage, thereby meeting stringent quality standards. In addition to its structural aspects, our study also examined the functional genomics of this species, including the intricate capacity to biosynthesize long-chain polyunsaturated fatty acids (LC-PUFAs) and secrete venom. Notably, our analyses revealed various repeats elements, which collectively constituted 17.43% of the genome. Moreover, annotation of 28,351 genes uncovered both shared genetic signatures and those that are unique to S. fuscescens. Our assembled genome also displayed a moderate prevalence of gene duplication compared to other fish species, which suggests that this species has a distinctive evolutionary trajectory and potentially unique functional constraints. Taken altogether, this genomic resource establishes a robust foundation for future research on the biology, evolution, and the aquaculture potential of S. fuscescens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Siganus fuscescens, also known as mottled spinefoot, is a tropical marine fish species belonging to the family Siganidae. It is widely distributed throughout the Indo-Pacific region, including the east coast of Africa, the Red Sea, the Persian Gulf, and the western Pacific Ocean (Woodland 1990). This species has also been reported in other areas in which it has not been known to occur, which is suggestive of its range expansion (Azzuro and Tiralongo 2020; Lenanton et al. 2017).
The mottled spinefoot is a small fish, reaching approximately 30 cm in length and has a distinctive, blue-spotted body pattern and a venomous spine on its dorsal fin that is used for predator defense. It inhabits shallow coral reef flats as well as algal and seagrass beds (Lieske and Myers 1994; Yamada et al. 1995). In terms of diet, S. fuscescens is primarily herbivorous and plays a significant ecological role in several marine ecosystems. It is known to feed predominantly on algae and, to a lesser extent, seagrass (Lieske and Myers 1994; Michael et al. 2013). One of the remarkable attributes of S. fuscescens is its ability to tolerate varied salinity levels, which enables it to inhabit regions characterized by fluctuating salinity, including mangroves (Duray 1998). This adaptability to variable salinity contributes to the ecological versatility of this species. Furthermore, S. fuscescens is also resilient to other environmental disturbances, which further enhances its ecological importance. The resilience has enabled S. fuscescens to withstand and recover from disturbances such as natural disasters and anthropogenic impacts (Cinner et al. 2013). Moreover, resilience demonstrated by S. fuscescens positions it as a valuable indicator species for assessing the health and ecological integrity of coral reef ecosystems (Zarco-Perello et al. 2021). This species holds significant cultural and culinary importance particularly in Southeast Asia and the Pacific Islands (Pertiwi et al. 2019; Wahyuningtyas et al. 2017) since it is highly regarded for its sweet, delicate flavor, and can be prepared using diverse cooking methods including grilling, steaming, and frying, indicating its versatility.
In recent years, there has been a growing interest in the use of S. fuscescens for aquaculture, primarily in Southeast Asian countries such as Indonesia and the Philippines (Jiarpinijnun et al. 2017; Li et al. 2018; Osako et al. 2006). This species possesses characteristics that make it suitable for aquaculture in coastal areas where other fish species may struggle. This is because S. fuscescens can be successfully raised in low-salinity and high-temperature environments that are prevalent in many parts of Southeast Asia. Siganus fuscescens also exhibits fast growth, high reproductive potential, and tolerance to a wide range of environmental conditions (Osako et al. 2006; Seale and Ellies 2019; Vergés et al. 2016). Even though such traits make it an attractive candidate for aquaculture, genomic information is required to identify the proper breeding and cultivation techniques which will support responsible and sustainable development of S. fuscescens aquaculture.
Therefore, in this study, we performed hybrid de novo genome assembly and annotation of S. fuscescens, and subsequently conducted comparative genomic analysis using ten other fish species representing diverse families. This is to provide a genomic resource as a means for an array of genetic, genomic, and phylogenetic studies of the mottled spinefoot and other fish species within the Siganidae family. In addition, our study also contributes to the exploitation of the potential economic value of S. fuscescens.
Materials and Methods
Specimen Collection and Preparation
The specimen under investigation was a live male fish labelled as “JG2022” which weighed 250 g and had a total length of 32.06 cm. It was captured off Jogashima Island in Japan by fishers and was subsequently transported to the Genome Science Laboratory of the Tokyo University of Marine Science and Technology (Tokyo, Japan). The transport container was filled with 20 L of oxygenated seawater to maintain optimal conditions.
The identification of the specimen was conducted using both meristic and morphometric characteristics. Meristic attributes were assessed via visual observation, following established guidelines for identification of Siganidae species (Woodland 2001). Morphometric attributes were assessed using standardized photographs of the specimen; photographs were analyzed using ImageJ version 1.53t (Schneider et al. 2012) and MorFishJ version 0.2.1 (Ghilardi 2022). Recorded attributes included body color, body shape, the number of dorsal fin spines and soft rays, the number of anal fin spines and soft rays, total length, standard length, body depth, eye diameter, and head length.
Finally, to collect samples, the specimen was first euthanized using an overdose of 0.5 mL/L 2-phenoxyethanol. A photograph of the specimen was then taken, providing a reference for future measurements. Ten organs, including the skin, muscle, liver, spleen, kidney, gut, testis, blood, gill, and brain, were harvested from the specimen, and stored at − 80 °C until use.
Nucleic Acid Extraction
Genomic DNA was extracted from collected tissues using a NucleoSpin® Tissue kit (Macherey-Nagel, Germany). The concentration and purity of all genomic DNA extracts were quantified using NanoDrop and Qubit 2.0 dsDNA BR Assay Kit (Invitrogen, USA) (Table S1—Supplementary File 1). In addition, an estimation of the molecular weight was generated using the visualization of DNA fragments on a 0.7% agarose gel stained with ethidium bromide. This was done to identify the most appropriate organ for obtaining high molecular weight DNA.
Library Preparation and Whole-Genome Sequencing
DNA extracted from muscle tissue was used for library preparation prior to sequencing. Two distinct sequencing platforms—i.e., Oxford Nanopore and Illumina—were used to generate sequence data. To generate long DNA reads, libraries were constructed utilizing an SQK-LSK109 ligation sequencing kit (Oxford Nanopore Technologies, UK) with a NEBNext companion module for ONT Ligation Sequencing (E7180, New England Biolabs). Subsequently, constructed libraries were sequenced on an ONT GridION-Mk1 platform, employing R9.4.1 flow cells. Base calling was performed using Guppy version 6.1.5 using the “super-accuracy” mode. In addition, quality filtering procedures were applied to all obtained data. To generate short DNA reads, paired-end libraries were prepared using a TruSeq DNA PCR Free (350) kit Illumina (Illumina, USA). These libraries were sequenced on an Illumina NovaSeq 6000 platform using an S4 flow cell (Eurofin Genomics, Tokyo).
Quality Checks and Filter-Trimming
The quality of raw reads from both ONT long read sequencing and Illumina short read sequencing was evaluated using FastQC version 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). NanoPlot version 1.40.2 (De Coster et al. 2018) was used to generate a histogram and density plot indicating the quality score and read length distribution for ONT reads. Illumina reads were first trimmed and filtered using fastp version 0.23.2 (Chen et al. 2018), while ONT reads were processed using Porechop_ABI version 0.5.0 (Bonenfant et al. 2022) and nanofilt version 2.8.0 (De Coster et al. 2018) to remove adapters and low-quality bases, respectively.
Genome Size Estimation
We then used Jellyfish version 2.2.10 to analyze the k-mer distribution of the trimmed long reads to get a preview of the genome composition and structure (Marçais and Kingsford 2011). A file containing the k-mer count histogram was then analyzed using GenomeScope version 2.0 (Ranallo-Benavidez et al. 2020) designed for k-mer-based analysis of genome characteristics (Vurture et al. 2017).
Mitogenome De Novo Assembly and Annotation
We first performed a de novo assembly of the S. fuscescens mitochondrial genome then annotated and visualized it using the MitoZ version 3.3 toolkit (Meng et al. 2019). Briefly, the pipeline implemented MEGAHIT version 1.0 (Li et al. 2016) with ‘kmer 59 79 99 119 141’ combination and annotation performed using the Chordata clade. To improve the quality of the annotation, the draft S. fuscescens mitogenome was reannotated using MitoAnnotator, a fish-specific tool sourced from the MitoFish version 3.85 mitogenomic database (http://mitofish.aori.u-tokyo.ac.jp/) (Sato et al. 2018; Iwasaki et al. 2013), accessed on January 10, 2023. Nucleotide BLAST of the assembled mitogenome was conducted on the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Subsequently, the Cytochrome c oxidase subunit I (COI) region of the mitogenome was queried against the Barcode of Life Database (BOLD) (https://www.boldsystems.org/). Both databases were accessed on January 10, 2023. Phylogenetic analysis was carried out using Siganidae species with complete mitogenomes retrieved from the NCBI database accessed on January 15, 2023. The analysis was performed using MAFFT version 7.471 (Katoh and Standley 2013) for sequence alignment and IQ-TREE version 2.2.0 (Minh et al. 2020) for phylogenetic tree construction using 1000 bootstrap replicates. S. fuscescens (accession number: EF025185.1) and Siganus canaliculatus (accession number: KJ872545.1) were used as the reference mitogenomes for this analysis.
Whole-Genome De Novo Assembly
We further conducted de novo assembly of the diploid genome using NextDenovo version 2.5.0 (Hu et al. 2023), a string graph-based assembler designed for long reads. We utilized Inspector version 1.2 (Chen et al. 2021) to identify and rectify any assembly errors present in the assembled genome. The quality of the assembled genome was assessed using Quast version 5.2 (Mikheenko et al. 2018) and the completeness of the genome was evaluated using Benchmarking Universal Single-copy Orthologs (BUSCO; version 5.4.3) (Manni et al. 2021) scores utilizing actinopterygii_odb10 lineage. Thereafter, we refined the draft assembly with Illumina short reads using NextPolish version 1.4.1 (Hu et al. 2020), a tool that is specifically tailored for NextDenovo-generated assemblies, then re-evaluated assembly statistics and genome completeness. At this stage, we had generated a diploid assembly. To reassign allelic contigs and eliminate haplotigs, we employed Purge_Haplotigs version 1.1.2 (Roach et al. 2018), a pipeline designed to identify pairs of contigs that are syntenic and move one of them to the haplotig ‘pool’ in genome assemblies. This pipeline utilizes mapped read coverage and Minimap2 alignments to determine which contigs to retain for the haploid assembly or phased-diploid assembly. The resulting haploid assembly statistics and completeness were evaluated using Quast and BUSCO scores, respectively, as described above.
Genome Annotation
Identification of De Novo Transposable Element Families
To identify repeats in our assembled genome, a de novo repeat library was first constructed using RepeatModeler version 2.0.5 (Chen 2004) applying RMBlast version 2.13.0 as the search engine. RepeatModeler is a software designed for identifying repeat families within genomes. It provides a list of repeat family sequences that can be utilized to mask repeats in a genome using tools like RepeatMasker. In the analysis, we integrated several tools for the structural analysis of long terminal repeats (LTR), including GenomeTools version 1.6.0 (Gremme et al. 2013), LTR-retriever version 2.9.0 (Ou and Jiang 2018), Ninja version 0.97-cluster_only, MAFFT version 7.471 (Katoh and Standley 2013), and CD-HIT version 4.8.1 (Fu et al. 2012). The resulting repeat consensus sequences were then refined by appending a species identifier to each consensus sequence ID. Finally, outcomes were categorized into two distinct groups: known and unknown repeat elements.
Repeat Annotation and Masking
The assembled S. fuscescens-JG2022 genome then underwent a soft masking process using RepeatMasker version 4.1.5 (Flynn et al. 2020). RepeatMasker is a widely used tool for identifying and masking repetitive elements in genomes. It acts as a wrapper around BLAST and other tools, utilizing sequence similarity with known repeats to identify and mask repetitive regions. The masking process in our study utilized species-specific repeat elements obtained from the step above and repeat elements of the teleost fish lineage sourced from Dfam version 3.7 database (Storer et al. 2021). The analysis was conducted sequentially starting with the identification and masking of species-specific repeats generated from this study then followed by lineage specific repeats carried out using RepeatModeler version 1.0.11 (Smit et al. 2015).
Gene Prediction and Functional Annotation
Gene prediction in this study was performed using BRAKER3 version 3.0.2 (Gabriel et al. 2023) with the default parameters. To improve the accuracy of these predictions, we harnessed protein-based evidence derived from the vertebrate subset of OrthoDB version 11 (Kuznetsov et al. 2023). BRAKER3 is a robust pipeline, adept at refining gene structure predictions by seamlessly integrating protein homology-based evidence to improve the precision of predictions (Brůna et al. 2020, 2021). This pipeline uses a two-step process: At the initial stage, the gene prediction tool AUGUSTUS was used to develop gene structure predictions (Hoff et al. 2016, 2019; Lomsadze et al. 2005), using the zebrafish (Danio rerio) gene model. The refinement process involved aligning forecasted gene structures and protein sequences sourced from the vertebrate division of OrthoDB version 11. This alignment was executed using DIAMOND version 0.9.24 and Spaln version 2.3.3d (Buchfink et al. 2015; Gotoh 2008; Iwata and Gotoh 2012) and enabled the inclusion of evolutionary relationship data, further improving the precision of gene predictions. Genomic features including number of coding sequences, introns, exons start, and stop codons from the predicted proteins were quantified using a custom script. Subsequently, the predicted proteins were blasted against the Swiss-Prot, TrEMBL, and the annotated proteins of nine fish species using DIAMOND version 2.1.8 for genes names assignment. Functional annotation of the predicted genes was performed using the eggNOG-mapper version 2 web server (Cantalapiedra et al. 2021). The amino acid sequences of predicted genes obtained from the BRAKER3 pipeline were used as an input for this analysis. The eggNOG-mapper employed the eggNOG 5.0 database (Huerta-Cepas et al. 2019) to assign functional annotations and orthology assignments to predicted genes, thereby leveraging evolutionary relationships. Annotations were transferred from orthologs using the Actinopterygii (7898) taxon scope to generate a taxa-specific annotation. This comprehensive analysis provided information on functional categories, orthologous groups, and domain predictions associated with our predicted genes. Gene Ontology (GO) enrichment analysis was conducted using the GO terms obtained from gene annotation. The analysis was performed using ClusterProfiler version 4.8.2 (Wu et al. 2021) in R version 4.3.0 (R Core Team 2023). All the annotation results were subjected to a filtration step where the longest transcripts with identity > 50% were retained for downstream analysis. We scanned the annotated genes for the presence of venom-related genes in fish (stonustoxin and neoverrucotoxin), long-chain polyunsaturated fatty acids (LC-PUFA) biosynthesis-related genes (fatty acid desaturase and elongation of very long fatty acid (ELOVL) elongase), and genes related to algae degradation (cellulolytic enzymes, agarases, carrageenases, and alginate lyase). These represented the hallmark features for S. fuscescens in relation to ecological adaptation.
Comparative Genomics
To investigate the evolution of gene families and to assess potential expansions or contractions in S. fuscescens-JG2022, we conducted comparative genomics analysis to capture phylogenetic relationships and evolution patterns. We expanded our analysis to include additional nine fish species: Anguilla anguilla, Chanos chanos, Danio rerio, Epinephelus fuscoguttatus, Gasterosteus aculeatus, Labrus bergylta, Salmo salar, Sebastes umbrosus, and Takifugu rubripes. Whole-genome sequences of these species were retrieved from NCBI (https://ftp.ncbi.nlm.nih.gov/genomes/refseq/vertebrate_other/) database (Table S2—Supplementary File 1). The selected fish species represent a diverse array of families, encompassing both well-studied model organisms like zebrafish, as well as species that exhibit unique traits relevant to our study. For instance, Atlantic salmon is renowned for its remarkable capacity to biosynthesize LC-PUFAs (Datsomor et al. 2022), while Ballan wrasse is a reef-associated fish species (Skiftesvik et al. 2015). This selection allowed us to capture a broad spectrum of adaptations and behaviors. BRAKER3 pipeline was used to generate gene predictions for all the species included in this study using uniform parameters as described earlier. The protein sequences generated were used as inputs for orthology analyses. OrthoFinder version 2.5.5 (Emms and Kelly 2015, 2017, 2019) with default settings was then used for detecting orthologous relationships. This algorithm utilizes reciprocal best hits and Markov clustering to categorize protein sequences into orthogroups based on sequence similarities and evolutionary relationships. The approach facilitated the exploration of orthologous relationships among genes, thereby painting a comprehensive picture of gene family dynamics across the analyzed species. We generated a maximum-likelihood (ML) phylogenetic tree by aligning concatenated proteins from single-copy orthologs using MAFFT version 7.471. FastTree version 2.1.11 (Price et al. 2010) was employed to construct the tree, utilizing the JTT + CAT evolution model (Lartillot and Philippe 2004) and performing a Shimodaira-Hasegawa (SH) test with 1000 bootstrap replicates to assess the confidence of the inferred splits (Shimodaira 2002). The divergence time between the species was estimated using TimeTree 5 (TimeTree :: The Timescale of Life) (Hedges et al. 2006). The median estimate for S. salar and A. anguilla was set at 224 million years ago. The gene family expansion and contraction were analyzed using CAFÉ version 5.1.0 (Mendes et al. 2020). Gene clusters comparison and visualization among the ten fish species and subsequently six species for Venn diagram construction was done using OrthoVenn3 applying default parameters (Sun et al. 2023).
Code Availability
The code utilized for data analysis in this study, along with the configurations and certain generated results from our study, is available through an online repository (https://github.com/Kishaz/Siganus_fuscescens-Genome_analysis_pipeline.git). Scripts for methods not provided here can be found in the original pipeline, with default settings as referenced.
Results
Morphological Identification
The morphometric measurements included in Table S3—Supplementary File 1 outline the distinct characteristics of the specimen. The fish displayed an olive-green body coloration contrasted with a silver belly, coupled with a sleek and compressed body structure. Notably, it featured thirteen dorsal spines, ten dorsal soft rays, seven anal spines, and nine anal soft rays, as illustrated in Fig. S1—Supplementary File 1. These features were consistent with those of S. fuscescens (Woodland 2001).
Whole-Genome Sequencing
We generated a total of 354,124,016 paired-end reads (150 bp each) from Illumina short read sequencing, amounting to 53.5 Gb in total. The raw reads had a GC content of 42.89% and a Q30 score of 86.24%. ONT long read sequencing yielded 4,842,762 reads, generating a total of 27.9 Gb, with a GC content of 42% and a Q12 score of 79.6% (Fig. S2—Supplementary File 1).
Genome Size Estimation
Genome size estimation yielded a range of heterozygosity values between 1.00712 and 1.00892%, denoting considerable genetic variation between the two haplotypes of S. fuscescens-JG2020. The haploid genome estimated size ranged from 500,030,638 to 500,164,805 bp (Fig. 1). The results here show a genome size that is smaller than the 567,000,000 bp (C-value, 0.58 pg) estimated genome size reported in the Animal Genome Size Database (Gregory 2002). The difference may be due to an additional parameter introduced in GenomeScope, set to exclude extremely high frequency k-mers as these likely represent organelle sequences or other contaminants that can inflate the genome size (Vurture et al. 2017). The underestimation of the genome size through sequence-based methods has been observed in other studies as reported by Pflug et al. (2020).

The k-mer distribution of ONT reads using GenomeScope based on a k value of 21. Genome haploid length (len): 500,164,805 bp; Unique (uniq): 90.2%; heterozygosity (het): 1.00892%; k-mer coverage (kcov): 17.8; Error rate (err): 1.91%; Read duplication (dup): 0.53%
Moreover, this analysis also indicated genome repeat lengths spanning from 48,782,197 to 48,795,286 bp, equivalent to approximately 9.754–9.756% of the estimated genome size. Conversely, the unique length of this genome was estimated to range between 451,248,441 and 451,369,519 bp, representing singularly occurring genome regions. The model fit, estimated by gauging the concordance between observed data and the anticipated distribution based on the genome model, ranged from 97.3422 to 98.2842%. These robust model fit values underscored a pronounced similarity between predicted and observed data, reinforcing the reliability of this analysis. In addition, the assessed read error rate was approximately 1.91107%. This value signified the percentage of sequencing errors within the acquired read dataset and necessitates the acknowledgment of errors that may translate in subsequent analysis from such errors.
Mitogenome De Novo Assembly and Annotation
Circular mitogenome of 16,494 bp was assembled using Illumina short reads. The average coverage depth of the mitogenome was determined to be 208-fold. Overall, the S. fuscescens-JG2022 mitogenome exhibited a GC content of 47%. Annotation of the assembled mitogenome generated thirteen protein-coding genes, two ribosomal RNA (rRNA) genes, and 22 transfer RNA (tRNA) genes. In addition, a control region was also identified in the mitogenome. Analysis of the gene distribution within the S. fuscescens-JG2022 mitogenome revealed that most genes were encoded on the heavy (H) strand. However, NADH dehydrogenase subunit 6 and the transfer RNAs for Gln, Ala, Asn, Cys, Tyr, Ser (UCN), Glu, and Pro were encoded on the light (L) strand. Finally, the architecture of the S. fuscescens-JG2022 mitochondrial genome was consistent with prior observations from other teleost species (Fig. 2).

De novo assembled Siganus fuscescens mitogenome using Illumina short reads
Nucleotide BLAST search of the whole mitogenome against the NCBI database revealed a 99.64% match to S. canaliculatus (accession number: KJ872545.1) and a 98.84% match to S. fuscescens (accession number: EF025185.1). Matching the Cytochrome c oxidase subunit I (COI) region against the BOLD database, specifically curated for the mitochondrial Cytochrome c oxidase subunit I gene, demonstrated a 100% identity of our sample to S. fuscescens. We conducted a phylogenetic analysis of S. fuscescens (sample JG2022) alongside other Siganidae species using publicly available complete mitogenomes. The analysis showed that S. fuscescens clustered closely with S. canaliculatus (Fig. S3 in Supplementary File 1). Multiple sequence alignment of the S. fuscescens-JG2020 mitogenome with S. fuscescens (accession number: EF025185.1) and S. canaliculatus (accession number: KJ872545.1) revealed high nucleotide similarity, with twelve nucleotide regions exhibiting variation (Fig. S4—Supplementary File 1).
Whole-Genome De Novo Assembly
Assembly Quality Metrics
The de novo assembly produced a total of 506 contigs with lengths ranging from 18,965 to 12,480,120 bp. The total length of the assembly was 494,613,806 bp and had a GC content of 42.89%. The N50 was 2,694,250 bp and the L50 was 59. After error correction and polishing, the number of contigs reduced to 179 and the length was 499,131,255 bp with a GC content slight increase to 42.92%. A drop in the number of contigs, from 179 to 164, was observed following curation using Purge_haplotigs. The final haploid genome had a length of 498,171,360 bp and a GC content of 42.92% (Table 1) and a summary of all the assembly statistics can be found in Table S4—Supplementary File 1. Importantly, the assembled genome accounted for 99.6% of the estimated genome size (~ 500 Mb).
Assessments of Assembly Completeness
Assessments of the completeness of the assembled genome were conducted be generating BUSCO scores at each step of the assembly process. The results revealed that NextDenovo assembly achieved a genome completeness of C:98.4%, while subsequent stages involving error correction and polishing demonstrated improved genome completeness scores of C:99.3% and C:99.5% respectively. The haplotype resolved assembly had a completeness score of C:99.5% (Fig. 3A). Error correction and polishing of the assembled genome had an overall positive effect on the completeness score. The analysis shows that the genome assembly is of high quality, containing almost all the expected single-copy genes with low duplication score.

A Genome completeness scores across different genome assembly and curation methods from BUSCO analysis using Actinopterygii_odb10 lineage. B Fraction of the assembled genome comprised of repeat elements in a stacked bar plot and the respective repeats landscape represented in a donut plot
Genome Annotation
Modelling and Annotation of Transposable Elements
During our genomic analysis of S. fuscescens-JG2022, we identified a total of 1116 de novo repeats entries, comprising 185 known and 931 putative elements. The 931 putative repeats elements might represent species-specific elements; however, we also acknowledge that they might exist in other organisms not covered in the reference database. Among the identified repeats elements, retroelements were the most prevalent, constituting 2.03% of the genome, followed closely by DNA transposons at 1.57%. Rolling-circle repeats accounted for 0.11%, while unclassified elements represented 6.69% of the repeat content. The remaining repeat categories included small RNAs (0.02%), simple repeats (6.1%), and low complexity sequences (0.88%) (Fig. 3B).
Gene Annotation
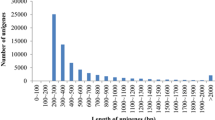
The distribution of the genomic features of the assembled genome of S. fuscescens-JG2022 is presented in Table 2. We identified a total of 77,149 predicted transcripts, encompassing 44,137 genes within the assembled genome. Our assessment revealed a BUSCO score of C:90.1% [S:89.3%, D:0.8%], F:3.1%, M:6.8%, n:3640, reflecting the overall completeness of our genomic annotation. However, it is important to note that the BUSCO score for predicted proteins was slightly lower than that of the whole genome. This difference could be attributed to the inherent limitations of gene prediction algorithms, which may not accurately capture all genes present in the genome. These findings highlight the complexities involved in genome annotation and highlight the ongoing efforts to refine computational methods for improved accuracy and completeness in genomic analyses. The average length of the predicted genes was approximately 6473 bp (Fig. S5—Supplementary File 1). Overall, we successfully annotated 28,351 of 44,137 predicted genes, encompassing 64.2% of the total. The protein annotation results and the GO information are summarized in Supplementary File 2. We identified several genes involved in the de novo biosynthesis of long-chain polyunsaturated fatty acids (LC-PUFAs). These genes included two copies of fatty acid desaturase 2 (Fads2) and a single copy of Fads6. Additionally, we detected members of the elongase family, including ELOVL-1, ELOVL-4, ELOVL-5, ELOVL-6, and ELOVL-7. Notably, the Fads2 genes were found to be multi-exonic and arranged in tandem within the genome. Further analysis revealed that the two Fads2 genes, Fads2a and Fads2b, exhibited sequence similarities of 93.21% and 85.81% with S. canaliculatus proteins B2KKL4.2 and D8 × 2C5.1, respectively. Based on the blast search results, Fads2a was designated as a Δ5/Δ6 desaturase, while Fads2b was designated as a Δ4/Δ5 desaturase. Our analysis revealed the presence of four venom-related genes, which we categorized into alpha and beta subunits. Among these, two specific genes, designated as sftoxin-a and sftoxin-b, exhibited significant similarities to known sequences from S. fuscescens (Kiriake et al. 2017). Specifically, sftoxin-a showed a high degree of similarity (99.71%) to the sftoxin-alpha subunit protein BAV10435.1, while sftoxin-b exhibited a similarity of 97.14% to the sftoxin-beta subunit protein BAV10436.1, both from S. fuscescens. The scan for genes related to algae degradation against the assembled genome of S. fuscescens-JG2022 returned no matches.
Comparative Genomics
Protein similarity analysis revealed that 12,820 orthogroups were shared across all the ten fish species while 8121 orthogroups, encompassing 19,959 genes, were specific to S. fuscescens (Fig. 4). For comparative genomic analysis, a total of 637,867 genes were generated from ten fish species, of which 80.9% were classified into orthogroups. Among the 56,169 orthogroups generated, 23,408 (41.7%) were species-specific, 12,820 orthogroups contained genes from all the ten fish species, and 339 orthogroups consisted of single-copy genes (Fig. 4). The analysis revealed that the fish species analyzed in our study had overlapping orthogroups which constituted shared genes suggesting conserved core biological functions.

Gene family cluster distribution, protein count, and intersections between Siganus fuscescens and nine other fish species. The green oval shapes interconnected using gray lines represent an UpSet plot which presents the number of orthologous clusters in each species, as well as the number of unique and shared homologous gene clusters among the study species. The dark green shade illustrates intersections while the light green shade represents data points that do not intersect
Our analysis indicates that S. fuscescens diverged from T. rubripes approximately 82.8 million years ago (MYA) (Fig. 5A). This pivotal divergence event coincided with a significant gene loss phenomenon, characterized by a net loss of 968 genes at this ancestral node. This observation highlights a critical juncture in the evolutionary trajectory of S. fuscescens, marked by substantial genomic reorganization and the depletion of genetic material. Analysis of gene gain and loss patterns revealed intriguing findings into the genomic evolution of S. fuscescens. Contrary to expectations, S. fuscescens exhibited the lowest level of gene expansion among the species examined, with a net gain of 192 genes (Fig. 5A). This contrasts with other fish species in our dataset, which displayed higher levels of gene expansion. The observed gene expansion in S. fuscescens suggests a more conservative genomic evolution, potentially driven by unique evolutionary constraints or ecological factors. Despite the lower gene expansion, S. fuscescens maintained a diverse gene repertoire essential for its biological functions and adaptation to its environment (Fig. 5B). Conversely, gene contraction events (−493) in S. fuscescens indicate a reduction in gene number compared to the reference state. While gene loss events are common across diverse taxa, the extent and nature of gene loss in S. fuscescens warrant further investigation to understand its implications for genomic and phenotypic evolution. It is important to note that the accuracy and completeness of the annotation process are influenced by the choice of analysis tools and the comprehensiveness of reference databases. Moreover, due to the substantial computational demands involved, we had to limit the number of fish species used for comparative genomic analysis. This constraint represents a significant limitation in our analytical approach.

A Phylogenetic analysis of Siganus fuscescens within the teleost lineage and the analysis of gene family gains (+) and losses (−). B Venn diagram illustrating shared and unique gene family clusters between six fish species
We examined the genes associated with venom secretion in fish and identified a diverse array. Siganus fuscescens harbored two distinct venom genes, discernible by their conserved domain structure. These genes were designated as the alpha and beta subunits, representing the major components of a potent dimeric venom protein. Phylogenetic analysis revealed a unique clade suggesting evolution of the gene to confer new function in specific fish species (Fig. S6—Supplementary File 1). The alpha subunit clade included S. fuscescens and S. umbrosus, both known for their ability to secrete venom, while the beta subunit clade encompassed almost all fish species, including non-venomous ones. Our investigation into the biosynthesis of long-chain polyunsaturated fatty acids (LC-PUFA) across the fish species under study unveiled a distinctive trait in S. fuscescens. Unlike other species, including S. salar, S. fuscescens had a significantly longer gene, almost double in length, comprising four domains. These domains included Cyt-b5, PLN03198 superfamily, Delta6-FADS-like, and gapped Delta6-FADS-like, all conserved within a single gene. In contrast, other species, such as Atlantic salmon, exhibited a split arrangement of these domains across multiple genes. Specifically, S. salar possessed separate genes for Cyt-b5 and Delta6-FADS-like, as well as for PLN03198 superfamily and gapped Delta6-FADS-like domains (Fig. 6).

Fatty acid desaturase–like gene tree representing a gene family derived from analysis of 10 fish species in this study. The conserved domains that have been in each gene have been illustrated adjacent the tree. The Siganus fuscescens Fads2 with bifunctional activity has been marked with a star. The conserved domains were searched in the NCBI database using Conserved Domains tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Delta6-FADS-like is Delta6 Fatty Acid Desaturase (Delta6-FADS)-like domain, Cyt-b5 is Cytochrome b5-like Heme/Steroid binding domain, and PLN03198 super family represents delta6-acyl-lipid desaturase. The characters after the species name represent the gene ID
Discussion
The morphological traits observed in our specimen align with those traditionally associated with S. fuscescens (Woodland 2001). However, ongoing challenges in correctly identifying S. fuscescens stem from its striking resemblance to a closely related sister species, S. canaliculatus (Park 1797). This similarity has complicated both research and conservation endeavors (Kuriiwa et al. 2007). Adding to the complexity is the suspicion of potential hybridization between these species, which further confounds identification issues. In certain research circles, there is a growing assertion that S. fuscescens and S. canaliculatus may indeed be indistinguishable and should be considered a single species (Hsu et al. 2011; Iwamoto et al. 2015). Our sample’s morphological traits closely resembled those of S. canaliculatus (Woodland 1997). Additionally, mitogenome analysis revealed slightly higher similarity to S. canaliculatus’ mitogenome compared to publicly available S. fuscescens reference mitogenome. This discrepancy likely arises from sequencing method differences: S. fuscescens’ mitogenome (accession number: EF025185.1) was sequenced via primer walking and Sanger sequencing, while S. canaliculatus’ (accession number: KJ872545.1) was sequenced using PCR-free NGS (Oh et al. 2007; Zhou et al. 2016). We found 12 nucleotide variations between our sample and the references, possibly due to amplification biases or mutations introduced during primer design. Limited reference mitogenomes highlight the need for broader nuclear genome markers. Our draft genome will make it possible to address this issue in future studies by incorporating diverse nuclear genome markers for enhanced resolution.
Genome annotation provided valuable information about the functional and structural attributes of S. fuscescens genes. Among the annotated genes, we identified homologs associated with specific adaptations of this species. These cover a range of functionalities, including, but not limited to, venom secretion (Kiriake et al. 2017), osmoregulation, pigment regulation, and the de novo biosynthesis of long-chain polyunsaturated fatty acids (LC-PUFA) (Osako et al. 2006). Our analysis revealed that S. fuscescens uses a unique biosynthesis pathway characterized by a bifunctional desaturase enzyme. Analysis of venom secretion–related genes revealed that its potent venom comprises two related subunits: the alpha and beta subunits as previously reported by Kiriake et al. (2017). Phylogenetic analysis suggested that the alpha subunit may have evolved from the beta subunit, which is conserved in most fish species studied, even those lacking venom-secreting abilities. Further examination revealed that both subunits share a conserved Thioredoxin_11 protein domain, with the beta subunit possessing an additional SPRY domain compared to the alpha subunit, which contains the SPRY_PRY_SNTX domain.
The synthesis of LC-PUFA in fish is essential for their adaptation to diverse habitats and has significant implications for aquaculture and consumer preferences (Ishikawa et al. 2019). Our investigation into S. fuscescens revealed the presence of two Fads2 genes encoding key desaturase enzymes responsible for LC-PUFA biosynthesis from precursor fatty acids, likely in response to dietary availability or specific physiological demands. Unlike other species with two-domain structures, S. fuscescens exhibited a unique gene structure with additional domains which confer bifunctional activity, suggesting a distinct evolutionary trajectory. The fatty acid desaturase had a unique structure with the presence of two Delta6 Fatty Acid Desaturase (Delta6-FADS)-like, one PLN03198 super family and one Cytochrome b5-like Heme/Steroid binding domains. In comparison, the other fish species only possessed two domains per gene, either Delta6-FADS-like with PLN03198 super family or Delta6-FADS-like with Cyt-b5 domain. Interestingly, the absence of ELOVL2, commonly found in other fish species, in the S. fuscescens genome highlights lineage-specific gene loss. However, the presence of ELOVL4 in S. fuscescens genome suggests a compensatory mechanism. Additionally, the detection of ELOVL8, a newly identified elongase member as reported by Li et al. (2020), emphasized the comprehensive repertoire of LC-PUFA biosynthesis genes in S. fuscescens (Li et al. 2020).
The results of our analysis were particularly intriguing since they indicated the absence of detectable genes—such as cellulolytic enzymes, agarases, carrageenases, and alginate lyase—that may be responsible for the digestion of algae- or plant-based material. This finding is noteworthy since algae serve as the primary diet of the study species (Avenant 2018). Recent research shows that most higher organisms cannot digest the cell wall in plant material without the assistance of microorganisms (Fujimori 2021; Zhu et al. 2018). We propose that S. fuscescens could potentially rely on symbiotic bacteria for the degradation of complex polysaccharides abundant in algae, facilitating the extraction of essential nutrients. This intriguing possibility highlights the need for further investigation into the microbial communities associated with S. fuscescens and their role in algae degradation and nutrient cycling within marine ecosystems.
Comparative genomic analysis of ten fish species, including S. fuscescens, revealed that a majority (80.9%) of their genes were allocated to orthogroups, which is indicative of robust species selection and aligns closely with the suggested > 80% threshold. These results indicate shared ancestry among the fish species, potentially evolving toward similar functionalities (Gabaldón and Koonin 2013). Notably, in the case of S. fuscescens-JG2022, 84.5% of its genes were assigned to orthogroups, leaving 15.5% unassigned which represent genes that were not found in the other nine fish species included in this study. The proportion of unassigned genes in S. fuscescens, may stem from multiple factors, including inadequate species sampling, gene duplication/loss, varied evolutionary rates, or incomplete gene annotation (Trachana et al. 2011). A subset of 19,545 genes were identified as belonging to species-specific orthogroups, a finding that is indicative of the acquisition of unique gene families specific to S. fuscescens. This phenomenon correlates with the observation of a net gain of 192 genes suggesting a selective pressure for the acquisition of novel genetic material, potentially facilitating adaptation to specific ecological niches or environmental challenges. Conversely, a considerable proportion of genes (11,946) remained unassigned, constituting about 15.5% of the total gene count. These unassigned genes may represent genetic relics of ancestral genes that are no longer functional or have undergone significant sequence divergence (Steinke et al. 2006).
Overall, our assembly and analysis of the S. fuscescens-JG2022 genome sheds light into its genetic composition and evolutionary history. The amalgamation of conserved gene families, species-specific genes, and unassigned genes highlights the complex interplay of gene expansion and contraction that has shaped the genomic landscape of S. fuscescens. Further investigation, including functional characterizations of unassigned genes and comparative analyses with related species, promises a deeper understanding of the genetic mechanisms underpinning unique characteristics and adaptive strategies of this species within the Siganidae family.
Conclusions
Our extensive genomic analysis of S. fuscescens-JG2022 has unveiled its genomic constitution and evolutionary context. We observed significant gene conservation, with 84.5% assigned to orthogroups, highlighting the presence of gene families shared with other fish species. A sizeable number of duplicate genes and the notable presence of species-specific genes suggest distinct evolutionary origins of many of these genes. Notably, the identification of genes associated with LC-PUFA carries significant implications for aquaculture. This discovery aligns with the increasing demand for LC-PUFA-rich fish, reducing reliance on external sources, addressing resource and environmental challenges, and providing health benefits. These findings improve our understanding of the evolutionary strategies of S. fuscescens within the Siganidae family. Furthermore, they also present opportunities for optimizing fish nutrition and creating sustainable aquaculture practices.
Data Availability
No datasets were generated or analyzed during the current study.
References
Avenant C (2018) Diet of the tropicalised herbivore S. fuscescens across a broad latitudinal gradient and comparisons with temperate seagrass-associated omnivorous fishes. Edith Cowan University. Retrieved from https://ro.ecu.edu.au/theses/2139
Azzuro E, Tiralongo F (2020) First record of the mottled spinefoot S. fuscescens (Houttuyn, 1782) in Mediterranean waters: a Facebook based detection. Mediterr Mar Sci 21:448–451
Bonenfant Q, Noé L, Touzet H (2023) Porechop_ABI: discovering unknown adapters in Oxford Nanopore Technology sequencing reads for downstream trimming. Bioinform Adv 3(1):vbac085. https://doi.org/10.1093/bioadv/vbac085
Brůna T, Lomsadze A, Borodovsky M (2020) GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform 2:lqaa026
Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M (2021) BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP + and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3:lqaa108
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J (2021) eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 38:5825–5829
Chen N (2004) Using repeat masker to identify repetitive elements in genomic sequences. Curr Protoc Bioinf 5:4–10
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890
Chen Y, Zhang Y, Wang AY, Gao M, Chong Z (2021) Accurate long read de novo assembly evaluation with Inspector. Genome Biol 22:1–21
Cinner JE, Huchery C, Darling ES, Humphries AT, Graham NA, Hicks CC, Marshall N, McClanahan TR (2013) Evaluating social and ecological vulnerability of coral reef fisheries to climate change. PLoS ONE 8:e74321
Datsomor AK, Gillard G, Jin Y, Olsen RE, Sandve SR (2022) Molecular regulation of biosynthesis of long chain polyunsaturated fatty acids in Atlantic salmon. Mar Biotechnol 24:661–670
De Coster W, D’hert S, Schultz DT, Cruts M, Van Broeckhoven C (2018) NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34:2666–2669
Duray MN (1998) Biology and culture of siganids. Aquaculture Department, Southeast Asian Fisheries Development Center
Emms DM, Kelly S (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16:157
Emms DM, Kelly S (2017) STRIDE: species tree root inference from gene duplication events. Mol Biol Evol 34:3267–3278
Emms DM, Kelly S (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20:238
Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF (2020) RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A 117:9451–9457
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152
Fujimori S (2021) Humans have intestinal bacteria that degrade the plant cell walls in herbivores. World J Gastroenterol 27:7784
Gabaldón T, Koonin EV (2013) Functional and evolutionary implications of gene orthology. Nat Rev Genet 14:360–366
Gabriel L, Brůna T, Hoff KJ, Ebel M, Lomsadze A, Borodovsky M, Stanke M (2023) BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiv. https://doi.org/10.1101/2023.06.10.544449
Ghilardi M (2022) MorFishJ: A software package for fish traditional morphometrics. Zenodo. https://doi.org/10.5281/zenodo.6969273
Gotoh O (2008) A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic Acids Res 36:2630–2638
Gregory TR (2002) Animal genome size database. https://www.genomesize.com/
Gremme G, Steinbiss S, Kurtz S (2013) GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE ACM Trans Comp Biol Bioinform 10:645–656
Hedges SB, Dudley J, Kumar S (2006) TimeTree: a public knowledgebase of divergence times among organisms. Bioinformatics 22:2971–2972
Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M (2016) BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32:767–769
Hoff KJ, Lomsadze A, Borodovsky M, Stanke M (2019) Whole-genome annotation with BRAKER. Gene Prediction. Humana, New York, NY, pp 65–95. https://doi.org/10.1007/978-1-4939-9173-0_5.
Hsu TH, Adiputra YT, Burridge CP, Gwo JC (2011) Two spinefoot colour morphs: mottled spinefoot S. fuscescens and white-spotted spinefoot S. Canaliculatus are synonyms. J Fish Biol 79:1350–1355
Hu J, Fan J, Sun Z, Liu S (2020) NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36:2253–2255
Hu J, Wang Z, Sun Z, Hu B, Ayoola AO, Liang F, Wang S (2024) NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biology 25(1):107. https://doi.org/10.1186/s13059-024-03252-4
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, von Mering C, Bork P (2019) eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47:D309–D314
Ishikawa A, Kabeya N, Ikeya K, Kakioka R, Cech JN, Osada N, Kitano J (2019) A key metabolic gene for recurrent freshwater colonization and radiation in fishes. Science 364:886–889
Iwamoto K, Abdullah MF, Chang CW, Yoshino T, Imai H (2015) Genetic isolation of the mottled spinefoot S. fuscescens ryukyu archipelago population. Biogeography 17:61–85
Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, Nishida M (2013) MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol 30:2531–2540
Iwata H, Gotoh O (2012) Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res 40:e161–e161
Jiarpinijnun A, Benjakul S, Pornphatdetaudom A, Shibata J, Okazaki E, Osako K (2017) High arachidonic acid levels in the tissues of herbivorous fish species (S. Fuscescens, Calotomus japonicus and Kyphosus bigibbus). Lipids 52:363–373
Katoh S, Standley K DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kiriake A, Ishizaki S, Nagashima Y, Shiomi K (2017) Occurrence of a stonefish toxin-like toxin in the venom of the rabbitfish S. fuscescens. Toxicon 140:139–146
Kuriiwa K, Hanzawa N, Yoshino T, Kimura S, Nishida M (2007) Phylogenetic relationships and natural hybridization in rabbitfishes (Teleostei: Siganidae) inferred from mitochondrial and nuclear DNA analyses. Mol Phylogenet Evol 45:69–80
Kuznetsov D, Tegenfeldt F, Manni M, Seppey M, Berkeley M, Kriventseva EV, Zdobnov EM (2023) OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res 51:D445–D451
Lartillot N, Philippe H (2004) A bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21:1095–1109
Lenanton RCJ, Dowling CE, Smith KA, Fairclough DV, Jackson G (2017) Potential influence of a marine heatwave on range extensions of tropical fishes in the eastern Indian Ocean—invaluable contributions from amateur observers. Reg Stud Mar Sci 13:19–31
Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Lam TW (2016) MEGAHIT v1. 0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102:3–11
Li Y, Zhang Q, Liu Y (2018) Rabbitfish–an emerging herbivorous marine aquaculture species. Aquaculture in China: success stories and modern trends. (pp 329–334). https://doi.org/10.1002/9781119120759.ch3_12
Li Y, Wen Z, You C, Xie Z, Tocher DR, Zhang Y, Li Y (2020) Genome wide identification and functional characterization of two LC-PUFA biosynthesis elongase (elovl8) genes in rabbitfish (Siganus canaliculatus). Aquaculture 522:735127
Lieske E, Myers R (1994) Collins pocket guide to coral reef fishes: IndoPacific and Caribbean. Herper Collins, London
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M (2005) Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 33:6494–6506
Manni M, Berkeley MR, Seppey M, Zdobnov EM (2021) BUSCO: assessing genomic data quality and beyond. Curr Protoc 1:e323
Marçais G, Kingsford C (2011) A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27:764–770
Mendes FK, Vanderpool D, Fulton B, Hahn MW (2020) CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36:5516–5518
Meng G, Li Y, Yang C, Liu S (2019) MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res 47:e63–e63
Michael PJ, Hyndes GA, Vanderklift MA, Vergés A (2013) Identity and behaviour of herbivorous fish influence large-scale spatial patterns of macroalgal herbivory in a coral reef. Mar Ecol Prog Ser 482:227–240
Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A (2018) Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34:i142–i150
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Oh DJ, Kim JY, Lee JA, Yoon WJ, Park SY, Jung YH (2007) Complete mitochondrial genome of the rabbitfish S. fuscescens (Perciformes, Siganidae) full length Research Paper. DNA Seq 18:295–301
Osako K, Saito H, Kuwahara K, Okamoto A (2006) Year-round high arachidonic acid levels in herbivorous rabbit fish S. fuscescens tissues. Lipids 41:473–489
Ou S, Jiang N (2018) LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol 176:1410–1422
Park M (1797) Descriptions of eight new fishes from Sumatra. Trans Linn Soc Lond 1:33–38. https://doi.org/10.1111/j.1096-3642.1797.tb00553.x
Pertiwi NPD, Hidayat NI, Henderson C, Putra ING, Sembiring A (2019) Redefining dispersal boundaries of S. fuscescens in the Coral Triangle area. Indo J Mar Sci 24:31-40. https://doi.org/10.14710/ik.ijms.24.1.31-40
Pflug JM, Holmes VR, Burrus C, Johnston JS, Maddison DR (2020) Measuring genome sizes using read-depth, k-mers, and flow cytometry: methodological comparisons in beetles (Coleoptera). G3: genes. Genomes Genet 10:3047–3060
Price MN, Dehal PS, Arkin AP (2010) FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490
R Core Team (2023) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org
Ranallo-Benavidez TR, Jaron KS, Schatz MC (2020) GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11:1432. https://www.nature.com/articles/s41467-020-14998-3. Accessed on 8 January 2023
Roach MJ, Schmidt SA, Borneman AR (2018) Purge haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19:460
Sato Y, Miya M, Fukunaga T, Sado T, Iwasaki W (2018) MitoFish and MiFish pipeline: a mitochondrial genome database of fish with an analysis pipeline for environmental DNA metabarcoding. Mol Biol Evol 35:1553–1555
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675
Seale AP, Ellies S (2019) Sustainable capture-based aquaculture of rabbitfish in Pacific Island lagoons. Aquacult Aquaponics 1:1–9
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Skiftesvik AB, Durif CM, Bjelland RM, Browman HI (2015) Distribution and habitat preferences of five species of wrasse (Family Labridae) in a Norwegian fjord. ICES J Mar Sci 72:890–899
Smit AFA, Hubley R, Green P (2015) RepeatMasker Open-4.0. 2013–2015. http://www.repeatmasker.org
Steinke D, Salzburger W, Braasch I, Meyer A (2006) Many genes in fish have species-specific asymmetric rates of molecular evolution. BMC Genomics 7:20
Storer J, Hubley R, Rosen J, Wheeler TJ, Smit AF (2021) The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob DNA 12:2
Sun J, Lu F, Luo Y, Bie L, Xu L, Wang Y (2023) OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res 51:W397–W403
Trachana K, Larsson TA, Powell S, Chen WH, Doerks T, Muller J, Bork P (2011) Orthology prediction methods: a quality assessment using curated protein families. BioEssays 33:769–780
Vergés A, Doropoulos C, Malcolm HA, Skye M, Garcia-Pizá M, Marzinelli EM, Campbell AH, Ballesteros E, Hoey AS, Vila-Concejo A, Bozec YM, Steinberg PD (2016) Long-term empirical evidence of ocean warming leading to tropicalization of fish communities, increased herbivory, and loss of kelp. Proc Natl Acad Sci U S A 113:13791–13796
Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, Schatz MC (2017) GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33:2202–2204
Wahyuningtyas LA, Nurilmala M, Sondita MFA, Taurusman AA, Sudrajat AO (2017) Nutritional profile of rabbitfish (Siganus spp.) from the kepulauan seribu (Thousand Islands), Jakarta, Indonesia. Int Food Res J 24:685
Woodland DJ (1990) Revision of the fish family Siganidae with descriptions of two new species and comments on distribution and biology. Indo-Pacific Fishes 19
Woodland D (1997) Siganidae. Spinefoots, rabbitfishes. FAO identification guide for fishery purposes. Western Cent Pac 3627–3650. http://www.fao.org/docrep/009/x2400e/x2400e00.htm
Woodland DJ (2001) Siganidae. In: Carpenter KE, Niem VH (eds) FAO species identification guide for fishery purposes. The living marine resources of the Western Central Pacific. Volume 6. Bony fishes part 4 (Labridae to Latimeriidae), estuarine crocodiles, sea turtles, sea snakes and marine mammal (PDF). FAO, Rome
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Yu G (2021) clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov 2(3). https://doi.org/10.1016/j.xinn.2021.100141
Yamada U, Shirai S, Irie T (1995) Names and illustrations of fishes from the East China Sea and the Yellow Sea. Japanese Chinese Korean
Zarco-Perello S, Bosch NE, Bennett S, Vanderklift MA, Wernberg T (2021) Persistence of tropical herbivores in temperate reefs constrains kelp resilience to cryptic habitats. J Ecol 109:2081–2094
Zhou L, Xie Z, Zhang Y (2016) The complete mitochondrial genome of the S. canaliculatus (Perciformes: Siganidae). Mitochondrial DNA Part A 27:1111–1112
Zhu L, Yang Z, Yao R, Xu L, Chen H, Gu X, Yang X (2018) Potential mechanism of detoxification of cyanide compounds by gut microbiomes of bamboo-eating pandas. MSphere 3:10–1128
Funding
This research was partly supported by the Science and Technology Research Partnership for Sustainable Development (SATREPS JPMJSA1806).
Author information
Authors and Affiliations
Contributions
Samuel Mwakisha Mwamburi, Satoshi Kawato, and Hidehiro Kondo were responsible for conceptualization. Samuel Mwakisha Mwamburi, Kayo Konishi, and Reiko Nozaki designed the methodology. Samuel Mwakisha Mwamburi, Satoshi Kawato, and Miho Furukawa performed formal analysis and data curation. Samuel Mwakisha Mwamburi was responsible for writing original draft and visualization. Satoshi Kawato, Hidehiro Kondo, and Ikuo Hirono participated in review and editing. Hidehiro Kondo supervised the project.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mwamburi, S.M., Kawato, S., Furukawa, M. et al. De Novo Assembly and Annotation of the Siganus fuscescens (Houttuyn, 1782) Genome: Marking a Pioneering Advance for the Siganidae Family. Mar Biotechnol (2024). https://doi.org/10.1007/s10126-024-10325-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10126-024-10325-9