
Abstract
Identifying quantitative trait loci (QTL) for viral disease resistance is of particular importance in selective breeding programs of fish species. Genetic markers linked to QTL can be useful in marker-assisted selection (MAS) for elites resistant to specific pathogens. Here, we conducted a genome scan for QTL associated with Singapore grouper iridovirus (SGIV) resistance in an Asian seabass (Lates calcarifer) family, using a high-density linkage map generated with genotyping-by-sequencing. One genome-wide significant and three suggestive QTL were detected at LG21, LG6, LG13, and LG15, respectively. The phenotypic variation explained (PVE) by the four QTL ranged from 7.5 to 15.6%. The position of the most significant QTL at LG21 was located between 31.88 and 36.81 cM. The SNP marker (SNP130416) nearest to the peak of this QTL was significantly associated with SGIV resistance in an unrelated multifamily population. One candidate gene, MECOM, close to the peak of this QTL region, was predicted. Evidence of alternative splicing was observed for MECOM and one specific category of splicing variants was differentially expressed at 5 days post-SGIV infection. The QTL detected in this study are valuable resources and can be used in the selective breeding programs of Asian seabass with regard to resistance to SGIV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aquaculture is becoming more and more important worldwide with the increasing demands for high-quality animal protein products (Yue 2014, Gjedrem et al. 2012). Frequent and extreme climate change and human-induced environmental pollutions also endanger natural fishery stocks and reduce capture production; thus, further increasing the demands for aquaculture products (Barange et al. 2014). Modern selective breeding programs based on marker-assisted selection (MAS) and the more advanced genomic selection (GS) are preferable methods to accelerate genetic improvement and overcome these challenges (Liu and Cordes 2004, Poompuang and Hallerman 1997, Yue and Wang 2017, Gjedrem et al. 2012). The molecular selective breeding strategies are being used in improving growth performance in fish species, e.g., salmon (Hershberger et al. 1990, Gjøen and Bentsen 1997), rainbow trout (Oncorhynchus mykiss) (Austreng et al. 1987), common carp (Cyprinus carpio) (Vandeputte 2003), large yellow croaker (Larimichthys crocea) (Dong et al. 2016), and Asian seabass (Lates calcarifer) (Yue 2014). Another economic trait, disease resistance, is also of particular importance and now has attracted more attention, because the outbreak of diseases is becoming more and more frequent and has caused severe mortality and great economic loss in the commercial stocks of almost all the fish species in the aquaculture industry (Fjalestad et al. 1993, Gjedrem 2015). One reason is that the commercial stocks suffer more from elevated stresses, e.g., stocking density, physiological stresses, and biochemical pollutions, in comparison to natural populations, and thus are susceptible to disease infections (Pickering and Pottinger 1989). However, most of the economic traits are complex traits and determined by quantitative trait loci (QTL), which significantly increases the difficulties of identifying the underlying genetic loci (Goddard and Hayes 2009). In contrast to the selection of growth performance, which can be achieved through traditional artificial selection, selection for viral and/or bacterial disease resistance would be more complicated and even problematic. Because the pathogen-challenging work can significantly increase the risks of infection among animals, the selection has to depend on the evaluation of the relatives of the challenged animals (Ødegård et al. 2011). Therefore, MAS and/or GS would be indispensable in the selective breeding programs for disease resistance traits (Ødegård et al. 2011).
To date, studies on identifying genetic loci associated with resistance to bacterial and viral diseases have been conducted in several fish species of great economic value, using linkage mapping. For instance, in Atlantic salmon (Salmo salar), a major QTL associated with resistance to infectious pancreatic necrosis virus (IPNV) was identified on linkage group 21 (Houston et al. 2010). In rainbow trout, QTL with relation to resistance to IPVN (Ozaki et al. 2001), infectious hematopoietic necrosis virus (IHNV) (Barroso et al. 2008), rhabdovirus (VHSV) (Verrier et al. 2013), hematopoietic necrosis (Rodriguez et al. 2004), and bacterial cold water disease (Johnson et al. 2008) were also detected and found to be distributed on diverse chromosomes. In Japanese flounder (Paralichthys olivaceus), a QTL of major effect on resistance to lymphocystis disease was detected by screening the resistant and susceptible individuals using linkage mapping (Fuji et al. 2006). Besides these QTL, several QTL associated with resistance to bacterial infection (Streptococcus iniae and Vibrio anguillarum) were also screened in this fish species (Wang et al. 2014a, Ozaki et al. 2010). These QTL, particularly those of major effect, have been used in the selective breeding programs to improve resistance to infection by specific pathogens of the commercial stocks (Chavanne et al. 2016).
Recent development of high-throughput marker discovery and genotyping driven by next-generation sequencing (Yue and Wang 2017) has significantly simplified the work in construction of high-density linkage maps and even genome-wide association studies (GWAS) in aquaculture species (Wang et al. 2015, Wang et al. 2017a, Wang et al. 2017b). Applications of these technologies have accelerated the processes of identifying loci underlying complex quantitative traits in selective breeding (Poland and Rife 2012). Recently, a number of studies have employed high-density genetic markers for QTL mapping and GWAS in aquaculture species. For example, a number of QTL related to growth, sexual maturation, and disease resistance have been identified in Atlantic salmon using GWAS (Gutierrez et al. 2015, Tsai et al. 2015, Correa et al. 2015). In catfish, a lot of QTL associated with disease resistance, stress tolerance, and growth performance were also screened using GWAS (Geng et al. 2015, Geng et al. 2016, Jin et al. 2017, Wang et al. 2017c). In some other aquaculture species that do not have available high-density SNP array, for example Asian seabass and Japanese flounder, genotyping-by-sequencing technology has been widely used for identifying loci with relation to growth and disease-resistant traits using QTL mapping and GWAS (Wang et al. 2015, Wang et al. 2017b, Liu et al. 2016b, Shao et al. 2015). It is suggested that applications of high-density genetic markers show superiority in power for mapping quantitative trait loci, even with minor effects (Liu et al. 2016b). In addition, such new technologies have also showed significant value of high efficiency and low cost, in the selective breeding of economic animals (De Donato et al. 2013).
Asian seabass is an important marine food fish species in Southeast Asian countries and Australia. The annual global production reached up to 75,000 tons in 2012 (Jerry 2013). Recently, the aquaculture industry of this species has been endangered by frequent outbreaks of viral diseases (Jerry 2013, de Groof et al. 2015). Among them, the disease caused by iridovirus infection is particularly severe (Gibson-Kueh et al. 2011). The pathogen, Singapore grouper iridovirus (SGIV), belonging to the genus Ranavirus of the Iridoviridae family, can infect a diverse range of vertebrate hosts and cause systemic diseases in many economically important fish species in the aquaculture industry, e.g., grouper, seabass, and sea bream (Qin et al. 2002). This virus can lead to a mortality rate of over 50% in fingerlings of these species (Qin et al. 2003). Although, the selective breeding programs of Asian seabass have been initiated since the early 2000s, most of the genetic improvements were focused on growth-related traits (Wang et al. 2006, Xia et al. 2013, Wang et al. 2015). To date, only some QTL with relation to resistance to viral nervous necrosis (VNN) disease have been identified, in our previous work (Liu et al. 2016b, Liu et al. 2016a). Therefore, identifying QTL, associated with resistance to SGIV infection, for MAS is indispensable in the mariculture industry of Asian seabass.
Here, we mapped QTL for resistance to iridovirus infection in Asian seabass, using genotyping-by-sequencing and linkage analysis. A high-density linkage map was constructed with 3261 markers including both microsatellites and SNPs. One genome-wide significant and three suggestive QTL for resistance to iridovirus infection were identified at four different linkage groups. The phenotypic variation explained by these QTL ranged from 7.5 to 15.6%. The SNP marker nearest to the peak of the most significant QTL region was significantly associated with SGIV resistance in an unrelated multifamily population. One candidate gene (MECOM) located in the most significant QTL that might be responsible for the phenotypic variation was identified. The expression of the gene was observed to be likely associated with SGIV infection. The identified QTL would be valuable resources for MAS in accelerating genetic improvements against SGIV infection in Asian seabass.
Materials and Methods
Animals and Experimental Design
The handling of animals in this study strictly followed the instructions of the Institutional Animal Care and Use Committee (IACUC) of Temasek Life Sciences Laboratory, Singapore (license no. TLL (F)-13-003). The animals used for virus challenge and QTL mapping were randomly selected from a F2 family. At 1-month post-hatch, the fish were moved from the Marine Aquaculture Center, Singapore, to Temasek Life Sciences Laboratory under laboratory condition. Approximately 2000 fish were cultured in four 100-l tanks with seawater. The seawater was maintained at 28 °C and with saturated oxygen and 30 ppt during the whole experiment. The tanks were constructed with a circulating water system and half of the water was replaced every day. Fish were fed twice every day with a common commercial diet (Marubeni Corporation, Japan) and were cultured in the facility for 2 weeks for acclimation before experimental challenge.
The Singapore grouper iridovirus (genus, Ranavirus) used for challenge was isolated from a brown-spotted grouper (Epinephelus tauvina) and was propagated in an embryonic cell line of the same species. The cells were cultured at 28 °C in Leibovitz’s L-15 media (Life Technologies, USA), with 10% fetal bovine serum (Life Technologies, USA) and 1% antibiotic antimycotic solution (Sigma-Aldrich, USA). The cells were harvested when SGIV produced a full cytopathic effect on the cells and were stored at −80 °C. The titer of SGIV was estimated using TCID50 (LaBarre and Lowy 2001). An immersion strategy was employed for virus challenge (Liu et al. 2016a). A concentration of 106 TCID50/ml of virus was used as determined based on the experience of our previous experimental tests. Fish from two randomly selected tanks were designated as the experimental groups and immersed into clean seawater with virus overnight. Fish from the other two tanks were designated as control groups and immersed into clean seawater with used L-15 media without virus. When a significant increase of mortality rate (>3 for two consecutive days) was observed in the infected fish, the dead fish were collected for QTL analysis. The experiment was stopped when the mortalities reached the standard line at which the number of fish deaths was <3 for two consecutive days (Liu et al. 2016a). The survivors were collected 1 week later. The traits were recorded in both binary (mortality vs. survival) and quantitative (days to death) format. The infection by SGIV in the fish was examined using a nested PCR-based method (Chao et al. 2002). The outer and inner primer sets are listed in Table S1. Genomic DNA was isolated using DNeasy Blood & Tissue Kits (Qiagen, Germany).
Genotyping of Genetic Markers
A double digest RADseq method (Peterson et al. 2012) was used for genotyping-by-sequencing, with some modifications, according to our previous work (Wang et al. 2016). Restriction enzymes PstI and MspI (New England Biolabs, USA) were selected for library construction. A total of three genotyping-by-sequencing (GBS) libraries were constructed for the 240 progeny and two parental fish. Two libraries, comprised of 192 progeny and the parental samples, had been sequenced using a NextSeq 500 platform (Illumina, USA) in a previous project, and 118 progeny were selected to construct a high-density genetic map for integration of the reference genome of Asian seabass (Wang et al. 2017a). In this study, the remaining one GBS library, comprised of the other 48 progeny, was prepared for single-end sequencing (1 × 150 bp) with the NextSeq 500 platform (Illumina, USA). All sequencing reads from the 240 progeny and two parents were used for downstream in silico SNP discovery and genotyping. The detailed information with regard to library construction and sequencing can be found in our previous study (Wang et al. 2017a). All the raw sequencing data have been archived in the DDBJ SRA database (Bioproject PRJDB5148).
Raw sequencing reads for each sample were filtered and trimmed to 130 bp using the program process_radtags implemented in the program package Stacks v1.42 (Catchen et al. 2011) with default parameters. Quality-controlled reads were aligned to the genome assembly of Asian seabass for SNP discovery, genotyping, and filtrations using Stacks v1.42 with the same parameters as our previous study (Wang et al. 2017a). The only difference was that the missing data cutoff and read depth were set to <20% and >7, respectively, in this study. Only one SNP was kept for each RAD tag for further analysis. Besides SNP markers, 95 microsatellites selected from our previous studies (Wang et al. 2017a, Liu et al. 2016a) were also genotyped across all the samples for QTL analysis.
Construction of a Genetic Map and Mapping of QTL for Resistance to Iridovirus
Genetic markers were filtered by examination of Mendelian segregation distortion using goodness-of-fit tests. Markers that significantly deviated from the Mendelian segregation at the level of 0.05 were removed from further analysis. Linkage map was constructed with the program JoinMap 4.1 (Van Ooijen 2006) with a LOD cutoff value of 10 and using the Kosambi mapping function of the regression mapping algorithm. QTL mapping was conducted using the program MapQTL6 (Van Ooijen and Kyazma 2009). Both interval mapping (IM) and multiple QTL model mapping (MQM) analyses were conducted. The LOD threshold value for determining each QTL was estimated by carrying out a permutation test for 1000 times. QTL with a LOD score more than the corresponding chromosome-wide and the genome-wide LOD threshold at the 0.05 level was considered as suggestive and significant, respectively. Both binary and quantitative traits data were used for QTL mapping.
QTL Verification and Candidate Gene Analysis in Multiple Families
Flanking sequences of the SNP nearest to the QTL peak region were retrieved from the reference genome of Asian seabass (Vij et al. 2016). This marker was genotyped using Sanger sequencing for QTL verification in a mass cross population, which was not genetically related to the mapping family (primer set Lca130416, Table S1). The population was generated by random crossing of 30 founders and was estimated to be comprised of at least 15 families. Challenge with SGIV was conducted using the above method for phenotyping. A total of 336 samples, i.e., 216 mortalities and 120 survivals, were collected for genotyping. Associations between genotypes and phenotypes were examined using Fisher’s exact test. Moreover, genomic sequences of the QTL region were retrieved to identify the most likely genes responsible for resistance to SGIV infection. Genes nearest to the most significant marker were selected as candidates. The expression profiles of the genes in six immune-related tissues, including the spleen, kidney, liver, intestine, skin, and brain, at 5 days post-challenge, in both the experimental and control groups, were further studied using Q-PCR. At least three individuals were used for each group. Q-PCR was performed on iQ™5 Real Time PCR Detection Systems (Bio-Rad, USA) using KAPA™ SYBR® Fast qPCR Kits (Kapa Biosystems, USA). We observed evidence of alternative splicing in one candidate gene. The relative expressions of the splicing variants were examined using Q-PCR. Primers were designed to be bridging exon/intron boundaries, so that the splicing variants could be differentiated. Total RNA isolation and cDNA synthesis were conducted according to our previous method (Wang et al. 2014b). The housekeeping gene elongation factor-1 alpha, EF1a (Table S1) was used as reference to characterize the relative gene expression using the 2−ΔΔCT method according to our previous study (Wang et al. 2014b).
Results
Virus Challenge and Genotyping-by-Sequencing
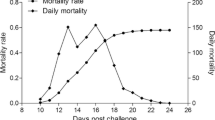
For the experimental groups, we observed that the mortality rate began to increase at 12 days post-challenge and later reduce to the background before the intensive mortalities appearing at 21 days post-challenge (Fig. 1). The overall mortality rate reached up to 45.1%. In comparison, the mortality rate in the control groups was smooth and low during the whole challenge experiment. Nested PCR examination revealed that all the fish in the experimental groups were infected by SGIV, while there were no virus infections in the controlled groups.

Daily mortality (left y-axis) of the experimental and control groups, and cumulative mortality rate (right y-axis) of the experimental groups over time
An average of 2.81 M raw reads were obtained for the 240 fish. After filtering out the samples with relatively low sequencing depth, 205 samples were retained, with an average of 3.12 M QC filtered reads per individual. The average number of QC filtered reads for each parent was 14.95 M. A catalog consisting of 133,199 loci that were identified from the RAD tags of the parents was used for SNP discovery and genotyping. A total of 4778 SNPs were genotyped in the mapping family. Moreover, all the 95 microsatellites were successfully genotyped in more than 95% of the individuals of the mapping family.
Linkage Map Construction
Among the 4873 genetic markers, 1532 significantly deviated from Mendelian segregation at the level of 0.05 and were therefore removed from further analysis. A total of 3341 markers including 3259 SNPs and 82 microsatellites were used for linkage mapping. A sex-averaged linkage map including 24 linkage groups (LGs) with a total length of 1358.29 cM was constructed with 3261 markers (Fig. 2 and Table S2). Summary statistics of the map and mapped markers are listed in Table 1. The length of each LG is from 15.67 cM for LG19 to 88.60 cM for LG12, with an average of 56.60 cM. The number of markers for each LG ranges from 20 in LG19 to 250 in LG2 with an average of 136, while the marker interval ranges from 0.26 cM in LG5 to 0.78 cM in LG19 with an average of 0.42 cM. The recombination rate in comparison to the physical map is from 0.82 cM/Mb for LG19 to 4.07 cM/Mb for LG23, with an average of 2.31 cM/Mb. The three largest intervals are in LG6, LG11, and LG15, with a length of 10.12, 9.06, and 8.99 cM, respectively (Fig. 2).

The sex-averaged linkage map and distribution of genetic markers along each linkage group in Asian seabass
QTL Mapping for Resistance to SGIV Infection and Verification
Genome scan of QTL for resistance to SGIV infection showed consistent results between IM and MQM mapping approaches. We also observed that the results, both in terms of the number of QTL and phenotypic variation explained (PVE), showed little difference between quantitative traits and binary traits (Table 2). QTL effects including LOD and PVE were slightly larger (~5%) for binary data than for quantitative data in three of the four QTL (LG6, LG13, and LG21) but smaller (~5%) for binary data in the remaining QTL (LG15). Here, we only report the results for binary data using interval mapping algorithm. A total of three suggestive and one genome-wide significant QTL were identified in LG6, LG13, LG15, and LG21, with a PVE of 7.5, 11.7, 11.2, and 15.6%, respectively (Table 2 and Fig. 3). The three suggestive QTL span a genomic region from 2.38 to 15.75 cM, while the genome-wide very significant QTL has an interval of 4.93 cM with a peak position at 36.81 cM in LG21.

LOD distributions along the sex-averaged linkage map for the whole genome scan for QTL in Asian seabass. The upper and lower dashed lines denote the LOD thresholds for estimated genome-wide significant (double asterisk) and suggestive (asterisk) QTL, respectively
The genetic marker (SNP130416) nearest to the peak of the most significant QTL in LG21 was further genotyped in an unrelated population (primer set: Lca130416, Table S1). We found significant differences in allele frequencies at this SNP between the mortality and the survival groups as examined using Fisher’s exact test (P = 0.04) (Fig. 4). Genotype CC was observed to be more common in the survival group (71.4%) than in the mortality group (60.0%). However, genotype CT was more abundant in the mortality group (37.8%) than in the survival group (28.6%).

Association between genotypes and phenotypes in an unrelated mass cross population of Asian seabass
Analysis of a Candidate Gene in QTL for Resistance to SGIV Infection
Genomic sequences with a length of 2.5 Mb flanking the genetic marker (SNP130416) nearest to the most significant QTL in LG21 were retrieved from the reference genome. We observed that this marker was located between two genes, the MDS1 and EVI1 complex locus (MECOM) and the sodium voltage-gated channel beta subunit 2 (SCN2B), and was ~23 and ~6 Kb away from the genomic sequences of the two genes, respectively (Fig. S1). The expression profiles of the two candidate genes at 5 days post-SGIV challenge were examined in six different immune-related tissues. The gene SCN2B showed no evidence of differential expression within each of the six tissues (primer set: Lca12931, Table S1). Interestingly, we observed evidence of splicing variants for the gene MECOM. Previous RNA-seq study revealed that MECOM has 18 exons, and alternative splicing occurs at four positions of three exons (5′-end of exon 10, 3′ and 5′-ends of exon 8, and exon 11 deletion), which produces 14 splicing variants in total (Vij et al. 2016). However, it was impossible to differentiate each splicing variant using Q-PCR, due to the random occurrence of the four alternative splicing positions (Fig. 5a). For this reason, we only examined the overall expression of MECOM (including all splicing variants) and the relative expression of each category of transcripts that showed splicing at each of the four alternative splicing positions (Fig. 5a). The transcripts were only found to be differentially expressed, upon the SGIV challenge, in the skin. The overall expression of the transcripts was significantly upregulated. For each category of transcripts, only the transcripts splicing with exon 11 deleted was observed to be significantly upregulated (Fig. 5b). Genomic sequences within the suggestive QTL regions were also retrieved and analyzed using the reference genome (Vij et al. 2016). Three candidate genes including membrane-associated guanylate kinase WW and PDZ domain-containing protein 1 (MAGI1), zinc transporter ZIP14 (ZIP14), and polymeric immunoglobulin receptor (PIGR), near to the peak region of each QTL in LG6, LG13, and LG15, respectively, were found to be most likely related to pathogen infection (Table 2).

Eighteen exons of the gene MECOM and the four alternative splicing sites within this gene in Asian seabass (a), and the overall expression of MECOM and the relative expression of each category of transcripts containing each of the four alternative splicing sites (b)
Discussion
To date, genotyping-by-sequencing has been widely used for construction of linkage maps and mapping QTL for economic traits in selective breeding programs of fish species (Gonen et al. 2014, Fu et al. 2016, Palaiokostas et al. 2013, Wang et al. 2015, Liu et al. 2015). This high throughput sequencing-based approach shows significant advances both in cost and labor, in comparison to traditional DNA markers that need to be genotyped by running gels (Peterson et al. 2012). In particular, using this approach, genotyping from several to dozens of thousands of SNPs for linkage mapping and QTL analysis is easily achievable. In most GBS-based studies, marker resolution can reach to <1 cM (Gonen et al. 2014, Palaiokostas et al. 2013, Wang et al. 2015, Fu et al. 2016), which is much higher than that in microsatellite-based studies (~5–10 cM) (Molina-Luzón et al. 2015, Negrín-Báez et al. 2016, Wang et al. 2011). In the present study, the length and resolution of linkage map are 1358.29 and 0.42 cM, respectively, which show little difference with the maps constructed using high-density SNP markers in our previous studies (length ~1350–1600 cM; resolution ~0.4–0.6 cM) (Wang et al. 2017a, Wang et al. 2015). This result supports confidence in this linkage map. The resolution of this map is also comparable to the studies in other fish species using GBS, e.g., in Atlantic salmon (~0.5 cM) (Lien et al. 2011), common carp (~0.4 cM) (Peng et al. 2016), bighead carp (~0.8 cM) (Fu et al. 2016), and large yellow croaker (~0.5 cM) (Ao et al. 2015). Therefore, this high-density linkage map can narrow down the QTL intervals and allow more accurate determination of the genomic region responsible for phenotypic variations, at a resolution of ~0.5 cM, corresponding to a genomic region of 0.24 Mb in Asian seabass (Vij et al. 2016).
QTL for resistance to viral and bacterial diseases are of particular importance in the selective breeding of both animal and plant species. These QTL can be used in MAS and incorporated into GS for improvement of disease resistance traits in brood stocks (Gjedrem 2015). Here, we identified four QTL associated with resistance to SGIV infection, a severe threat to the mariculture industry of Asian seabass in the Asia Pacific region (Qin et al. 2003). As far as we know, this is the first study in mapping QTL for iridovirus resistance in Asian seabass. Interestingly, we found little difference between quantitative and binary traits in the detection of QTL for disease resistance. This was also observed in the study of Atlantic salmon for identifying QTL for resistance to Piscirickettsia salmonis disease (Correa et al. 2015), supporting confidence in our QTL mapping results. The PVE for these four QTL ranges from 7.5 to 15.6%. In comparison to the major QTL identified in some other studies, e.g., for resistance to freshwater IPN in Atlantic salmon (PVE, ~50%) (Houston et al. 2010), resistance to viral lymphocystis disease in Japanese flounder (PVE, ~50%) (Fuji et al. 2006) and for bacterial disease resistance in rainbow trout (PVE, ~50–86%) (Baerwald et al. 2011), the effects of the QTL identified in this study are relatively small, although one of them was revealed to be genome-wide very significant. These results likely suggest that resistance to SGIV in Asian seabass is a complex polygenic trait and controlled by multiple genes of only small effects (Wang et al. 2017b). It is also likely that there is no major QTL for resistance to this specific pathogen, at least in the studied population.
Most of the QTL studies in fish species have revealed that resistance to viral and bacterial disease traits are controlled by multiple genetic loci, and each QTL usually has only minor effect (PVE, ~10%) (Liu et al. 2016a, Massault et al. 2011, Rodríguez-Ramilo et al. 2013, Geng et al. 2015, Wang et al. 2017b). However, it should be noted that these studies are limited by the effective size of the mapping population. In particular, most of the studies are based on only one biparental population. It is likely that the corresponding mutation responsible for resistance to a specific pathogen is missing in the mapping population. Even in genome-wide association studies (GWAS) of aquaculture fish species, the effective sizes of mapping populations are also rather small compared to natural populations, as the founder populations are usually set up by capturing dozens of wild individuals (Kincaid 1983). Thus, it is also difficult to identify QTL of major effects using GWAS in these species (Geng et al. 2015, Correa et al. 2015). In natural populations, genetic variations associated with pathogen resistance are maintained in the host-pathogen coevolutionary interactions (Anderson and May 1982), e.g., MHC polymorphisms under selection of pathogens (Borghans et al. 2004). Thus, there should be specific mutations for a host in response to a specific pathogen infection (Moen et al. 2015). It is very likely that the resistance alleles have minor frequencies and are therefore not included in the brooding stocks or the mapping families (McKay and Latta 2002). Although application of a single family can improve the power of detection of QTL, it is also prone to fail in capturing the mutations of major effects on a specific trait (Liu et al. 2016a). For this reason, QTL studies based on multiple families and GWAS using populations of large effective size are necessary to identify genetic loci of common and major effects (Sneller et al. 2009). Nevertheless, the marker (SNP130416) within the genome-wide significant QTL was revealed to be significantly associated with SGIV resistance in an unrelated mass cross population. This marker is of particular importance for MAS in the brood stocks of Asian seabass. Interestingly, the homozygous and heterozygous genotypes, CC and CT, were abundant in the survival and mortality groups, respectively, which might suggest that allele C is a recessive resistance allele. Moreover, all the QTL identified in the study are also useful in GS for disease resistance and robustness based on thousands of selected markers of diverse QTL effects (Goddard and Hayes 2007).
Within the genomic region of the most significant QTL in this study, two candidate genes were identified nearest to the peak of this QTL interval. However, only one gene, MECOM, was observed to be differentially expressed in response to infection by SGIV, although it is more than 20 Kb away from the marker. Interestingly, MECOM was found to be upregulated in the skin, but not in the main immune tissues or organs, e.g., kidney, spleen, and liver. MECOM has been widely studied and plays important roles in viral responses (Métais and Dunbar 2008). The protein coded contains a viral integration site and has a DNA-binding activity. As a transcription factor, this protein can induce various downstream signaling pathways related to disease responses (Sood et al. 1999). However, it is still not clear how this gene responds to SGIV infection in Asian seabass. Taking into account that MECOM showed evidence of alternative splicing in the face of infection, future studies should focus on the mechanism underlying the interactions between alternative splicing and virus infection, and the potential functions of each splicing variant. However, it should be noted that the other genes within the most significant QTL region are also likely to be causative genes. The QTL region has a length of ~2.5 Mb and covers dozens of genes according to the annotation of the Asian seabass reference genome (Vij et al. 2016). Due to linkage and disequilibrium, accurate determination of the causative genes in the studied biparental population is unlikely. Thus, we cannot exclude the possibilities that the other genes within this QTL region are the causative genes for resistance to iridovirus in Asian seabass. It is necessary to accurately determine the causative genes using association mapping based on a population of large effective size in the near future.
Conclusion
In this study, a high-density linkage map was constructed based on 3261 SNPs and microsatellites. This map was used for mapping QTL for resistance to SGIV infection in Asian seabass. Three suggestive and one genome-wide significant QTL were detected at four different chromosomes. The PVE for these QTL ranges from 7.5 to 15.6%. The genetic marker nearest to the most significant QTL was revealed to be significantly associated with SGIV resistance in an unrelated population of Asian seabass. The QTL identified in the study could be useful both for MAS and future GS in the selective breeding programs of Asian seabass. A candidate gene, MECOM, was detected at chromosome 21. Further study on the potential function of this gene can provide important information for understanding the mechanism of host-pathogen interaction and resistance to SGIV in Asian seabass.
References
Anderson RM, May R (1982) Coevolution of hosts and parasites. Parasitology 85:411–426
Ao J, Li J, You X, Mu Y, Ding Y, Mao K, Bian C, Mu P, Shi Q, Chen X (2015) Construction of the high-density genetic linkage map and chromosome map of large yellow croaker (Larimichthys crocea). Int J Mol Sci 16:26237–26248
Austreng E, Storebakken T, Åsgård T (1987) Growth rate estimates for cultured Atlantic salmon and rainbow trout. Aquaculture 60:157–160
Baerwald M, Petersen J, Hedrick R, Schisler G, May B (2011) A major effect quantitative trait locus for whirling disease resistance identified in rainbow trout (Oncorhynchus mykiss). Heredity 106:920–926
Barange M, Merino G, Blanchard J, Scholtens J, Harle J, Allison E, Allen J, Holt J, Jennings S (2014) Impacts of climate change on marine ecosystem production in societies dependent on fisheries. Nat Clim Chang 4:211–216
Barroso RM, Wheeler PA, Lapatra SE, Drew RE, Thorgaard GH (2008) QTL for IHNV resistance and growth identified in a rainbow (Oncorhynchus mykiss)× Yellowstone cutthroat (Oncorhynchus clarki bouvieri) trout cross. Aquaculture 277:156–163
Borghans JA, Beltman JB, De Boer RJ (2004) MHC polymorphism under host-pathogen coevolution. Immunogenetics 55:732–739
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks: building and genotyping loci de novo from short-read sequences. G3 (Bethesda) 1:171–182
Chao C, Yang S, Tsai H, Chen C, Lin C, Huang H (2002) A nested PCR for the detection of grouper iridovirus in Taiwan (TGIV) in cultured hybrid grouper, giant seaperch, and largemouth bass. J Aquat Anim Health 14:104–113
Chavanne H, Janssen K, Hofherr J, Contini F, Haffray P, Komen H, Nielsen EE, Bargelloni L, Consortium A (2016) A comprehensive survey on selective breeding programs and seed market in the European aquaculture fish industry. Aquac Int 24:1287–1307
Correa K, Lhorente JP, López ME, Bassini L, Naswa S, Deeb N, Di Genova A, Maass A, Davidson WS, Yáñez JM (2015) Genome-wide association analysis reveals loci associated with resistance against Piscirickettsia salmonis in two Atlantic salmon (Salmo salar L.) chromosomes. BMC Genomics 16:854
De Donato M, Peters SO, Mitchell SE, Hussain T, Imumorin IG (2013) Genotyping-by-sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One 8:e62137
De Groof A, Guelen L, Deijs M, Van Der Wal Y, Miyata M, Ng KS, Van Grinsven L, Simmelink B, Biermann Y, Grisez L (2015) A novel virus causes scale drop disease in Lates calcarifer. PLoS Pathog 11:e1005074
Dong L, Xiao S, Chen J, Wan L, Wang Z (2016) Genomic selection using extreme phenotypes and pre-selection of SNPs in large yellow croaker (Larimichthys crocea). Mar Biotechnol 18:575–583
Fjalestad KT, Gjedrem T, Gjerde B (1993) Genetic improvement of disease resistance in fish: an overview. Aquaculture 111:65–74
Fu B, Liu H, Yu X, Tong J (2016) A high-density genetic map and growth related QTL mapping in bighead carp (Hypophthalmichthys nobilis). Sci Rep 6:28679
Fuji K, Kobayashi K, Hasegawa O, Coimbra MRM, Sakamoto T, Okamoto N (2006) Identification of a single major genetic locus controlling the resistance to lymphocystis disease in Japanese flounder (Paralichthys olivaceus). Aquaculture 254:203–210
Geng X, Liu S, Yao J, Bao L, Zhang J, Li C, Wang R, Sha J, Zeng P, Zhi D (2016) A genome-wide association study identifies multiple regions associated with head size in catfish. G3 (Bethesda) 6:3389–3398
Geng X, Sha J, Liu S, Bao L, Zhang J, Wang R, Yao J, Li C, Feng J, Sun F (2015) A genome-wide association study in catfish reveals the presence of functional hubs of related genes within QTLs for columnaris disease resistance. BMC Genomics 16:196
Gibson-Kueh S, Thuy N, Elliot A, Jones J, Nicholls P, Thompson R (2011) An intestinal Eimeria infection in juvenile Asian seabass (Lates calcarifer) cultured in Vietnam—a first report. Vet Parasitol 181:106–112
Gjedrem T (2015) Disease resistant fish and shellfish are within reach: a review. J Mar Sci Eng 3:146–153
Gjedrem T, Robinson N, Rye M (2012) The importance of selective breeding in aquaculture to meet future demands for animal protein: a review. Aquaculture 350:117–129
Gjøen H, Bentsen H (1997) Past, present, and future of genetic improvement in salmon aquaculture. ICES J Mar Sci 54:1009–1014
Goddard ME, Hayes B (2007) Genomic selection. J Anim Breed Genet 124:323–330
Goddard ME, Hayes BJ (2009) Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet 10:381–391
Gonen S, Lowe NR, Cezard T, Gharbi K, Bishop SC, Houston RD (2014) Linkage maps of the Atlantic salmon (Salmo salar) genome derived from RAD sequencing. BMC Genomics 15:166
Gutierrez AP, Yáñez JM, Fukui S, Swift B, Davidson WS (2015) Genome-wide association study (GWAS) for growth rate and age at sexual maturation in Atlantic salmon (Salmo salar). PLoS One 10:e0119730
Hershberger W, Myers J, Iwamoto R, Mcauley W, Saxton A (1990) Genetic changes in the growth of coho salmon (Oncorhynchus kisutch) in marine net-pens, produced by ten years of selection. Aquaculture 85:187–197
Houston RD, Haley CS, Hamilton A, Guy DR, Mota-Velasco JC, Gheyas AA, Tinch AE, Taggart J, Bron J, Starkey W (2010) The susceptibility of Atlantic salmon fry to freshwater infectious pancreatic necrosis is largely explained by a major QTL. Heredity 105:318–327
Jerry DR (2013) Biology and culture of Asian seabass Lates calcarifer. CRC Press, New York
Jin Y, Zhou T, Geng X, Liu S, Chen A, Yao J, Jiang C, Tan S, Su B, Liu Z (2017) A genome-wide association study of heat stress-associated SNPs in catfish. Anim Genet 48:233–236
Johnson NA, Vallejo RL, Silverstein JT, Welch TJ, Wiens GD, Hallerman EM, Palti Y (2008) Suggestive association of major histocompatibility IB genetic markers with resistance to bacterial cold water disease in rainbow trout (Oncorhynchus mykiss). Mar Biotechnol 10:429–437
Kincaid HL (1983) Inbreeding in fish populations used for aquaculture. Aquaculture 33:215–227
Labarre DD, Lowy RJ (2001) Improvements in methods for calculating virus titer estimates from TCID 50 and plaque assays. J Virol Methods 96:107–126
Lien S, Gidskehaug L, Moen T, Hayes BJ, Berg PR, Davidson WS, Omholt SW, Kent MP (2011) A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics 12:615
Liu P, Wang L, Wan ZY, Ye BQ, Huang S, Wong S-M, Yue GH (2016a) Mapping QTL for resistance against viral nervous necrosis disease in Asian seabass. Mar Biotechnol 18:107–116
Liu P, Wang L, Wong S-M, Yue GH (2016b) Fine mapping QTL for resistance to VNN disease using a high-density linkage map in Asian seabass. Sci Rep 6:32122
Liu S, Vallejo RL, Gao G, Palti Y, Weber GM, Hernandez A, Rexroad Iii CE (2015) Identification of single-nucleotide polymorphism markers associated with cortisol response to crowding in rainbow trout. Mar Biotechnol 17:328–337
Liu Z, Cordes J (2004) DNA marker technologies and their applications in aquaculture genetics. Aquaculture 238:1–37
Massault C, Franch R, Haley C, De Koning D, Bovenhuis H, Pellizzari C, Patarnello T, Bargelloni L (2011) Quantitative trait loci for resistance to fish pasteurellosis in gilthead sea bream (Sparus aurata). Anim Genet 42:191–203
Mckay JK, Latta RG (2002) Adaptive population divergence: markers, QTL and traits. Trends Ecol Evol 17:285–291
Métais J-Y, Dunbar CE (2008) The MDS1–EVI1 gene complex as a retrovirus integration site: impact on behavior of hematopoietic cells and implications for gene therapy. Mol Ther 16:439–449
Moen T, Torgersen J, Santi N, Davidson WS, Baranski M, Ødegård J, Kjøglum S, Velle B, Kent M, Lubieniecki KP (2015) Epithelial cadherin determines resistance to infectious pancreatic necrosis virus in Atlantic salmon. Genetics 200:1313–1326
Molina-Luzón MJ, Hermida M, Navajas-Pérez R, Robles F, Navas JI, Ruiz-Rejón C, Bouza C, Martínez P, De La Herrán R (2015) First haploid genetic map based on microsatellite markers in Senegalese sole (Solea senegalensis, Kaup 1858). Mar Biotechnol 17:8–22
Negrín-Báez D, Navarro A, Afonso J, Toro M, Zamorano M (2016) Quantitative trait loci for a neurocranium deformity, lack of operculum, in gilthead seabream (Sparus aurata L.) Anim Genet 47:230–233
Ødegård J, Baranski M, Gjerde B, Gjedrem T (2011) Methodology for genetic evaluation of disease resistance in aquaculture species: challenges and future prospects. Aquac Res 42:103–114
Ozaki A, Okamoto H, Yamada T, Matuyama T, Sakai T, Fuji K, Sakamoto T, Okamoto N, Yoshida K, Hatori K (2010) Linkage analysis of resistance to streptococcus iniae infection in Japanese flounder (Paralichthys olivaceus). Aquaculture 308:S62–S67
Ozaki A, Sakamoto T, Khoo S, Nakamura K, Coimbra M, Akutsu T, Okamoto N (2001) Quantitative trait loci (QTLs) associated with resistance/susceptibility to infectious pancreatic necrosis virus (IPNV) in rainbow trout (Oncorhynchus mykiss). Mol Gen Genom 265:23–31
Palaiokostas C, Bekaert M, Davie A, Cowan ME, Oral M, Taggart JB, Gharbi K, Mcandrew BJ, Penman DJ, Migaud H (2013) Mapping the sex determination locus in the Atlantic halibut (Hippoglossus hippoglossus) using RAD sequencing. BMC Genomics 14:566
Peng W, Xu J, Zhang Y, Feng J, Dong C, Jiang L, Feng J, Chen B, Gong Y, Chen L (2016) An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio). Sci Rep 6:26693
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7:e37135
Pickering A, Pottinger T (1989) Stress responses and disease resistance in salmonid fish: effects of chronic elevation of plasma cortisol. Fish Physiol Biochem 7:253–258
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5:92–102
Poompuang S, Hallerman EM (1997) Toward detection of quantitative trait loci and marker-assisted selection in fish. Rev Fish Sci 5:253–277
Qin Q, Chang S, Ngoh-Lim G, Gibson-Kueh S, Shi C, Lam T (2003) Characterization of a novel ranavirus isolated from grouper Epinephelus tauvina. Dis Aquat Org 53:1–9
Qin QW, Shi C, Gin KY, Lam TJ (2002) Antigenic characterization of a marine fish iridovirus from grouper, Epinephelus spp. J Virol Methods 106:89–96
Rodríguez-Ramilo S, Fernández J, Toro M, Bouza C, Hermida M, Fernández C, Pardo B, Cabaleiro S, Martínez P (2013) Uncovering QTL for resistance and survival time to Philasterides dicentrarchi in turbot (Scophthalmus maximus). Anim Genet 44:149–157
Rodriguez MF, Lapatra S, Williams S, Famula T, May B (2004) Genetic markers associated with resistance to infectious hematopoietic necrosis in rainbow and steelhead trout (Oncorhynchus mykiss) backcrosses. Aquaculture 241:93–115
Shao C, Niu Y, Rastas P, Liu Y, Xie Z, Li H, Wang L, Jiang Y, Tai S, Tian Y (2015) Genome-wide SNP identification for the construction of a high-resolution genetic map of Japanese flounder (Paralichthys olivaceus): applications to QTL mapping of Vibrio anguillarum disease resistance and comparative genomic analysis. DNA Res 22:161–170
Sneller C, Mather D, Crepieux S (2009) Analytical approaches and population types for finding and utilizing QTL in complex plant populations. Crop Sci 49:363–380
Sood R, Talwar-Trikha A, Chakrabarti S, Nucifora G (1999) MDS1/EVI1 enhances TGF-β1 signaling and strengthens its growth-inhibitory effect, but the leukemia-associated fusion protein AML1/MDS1/EVI1, product of the t (3; 21), abrogates growth-inhibition in response to TGF-β1. Leukemia 13:348–357
Tsai H-Y, Hamilton A, Tinch AE, Guy DR, Gharbi K, Stear MJ, Matika O, Bishop SC, Houston RD (2015) Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genomics 16:969
Van Ooijen J (2006) JoinMap® 4, software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Van Ooijen J, Kyazma B (2009) MapQTL 6® 4, software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma BV, Wageningen
Vandeputte M (2003) Selective breeding of quantitative traits in the common carp (Cyprinus carpio): a review. Aquat Living Resour 16:399–407
Verrier ER, Dorson M, Mauger S, Torhy C, Ciobotaru C, Hervet C, Dechamp N, Genet C, Boudinot P, Quillet E (2013) Resistance to a rhabdovirus (VHSV) in rainbow trout: identification of a major QTL related to innate mechanisms. PLoS One 8:e55302
Vij S, Kuhl H, Kuznetsova IS, Komissarov A, Yurchenko AA, Van Heusden P, Singh S, Thevasagayam NM, Prakki SRS, Purushothaman K (2016) Chromosomal-level assembly of the Asian seabass genome using long sequence reads and multi-layered scaffolding. PLoS Genet 12:e1005954
Wang CM, Bai ZY, He XP, Lin G, Xia JH, Sun F, Lo LC, Feng F, Zhu ZY, Yue GH (2011) A high-resolution linkage map for comparative genome analysis and QTL fine mapping in Asian seabass, Lates calcarifer. BMC Genomics 12:174
Wang CM, Lo LC, Zhu ZY, Yue GH (2006) A genome scan for quantitative trait loci affecting growth-related traits in an F1 family of Asian seabass (Lates calcarifer). BMC Genomics 7:274
Wang L, Bai B, Liu P, Huang SQ, Wan ZY, Chua E, Ye B, Yue GH (2017a) Construction of high-resolution recombination maps in Asian seabass. BMC Genomics 18:63
Wang L, Fan C, Liu Y, Zhang Y, Liu S, Sun D, Deng H, Xu Y, Tian Y, Liao X, Xie M, Li W, Chen S (2014a) A genome scan for quantitative trait loci associated with Vibrio anguillarum infection resistance in Japanese flounder (Paralichthys olivaceus) by bulked segregant analysis. Mar Biotechnol 16:513–521
Wang L, Liu P, Huang S, Ye B, Chua E, Wan ZY, Yue GH (2017b) Genome-wide association study identifies loci associated with resistance to viral nervous necrosis disease in Asian seabass. Mar Biotechnol 19:255–265
Wang L, Wan ZY, Bai B, Huang SQ, Chua E, Lee M, Pang HY, Wen YF, Liu P, Liu F (2015) Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Sci Rep 5:16358
Wang L, Wan ZY, Lim HS, Yue GH (2016) Genetic variability, local selection and demographic history: genomic evidence of evolving towards allopatric speciation in Asian seabass. Mol Ecol 25:3605–3621
Wang L, Xia JH, Liu XJ, Liu P, Wan ZY, Yue GH (2014b) Molecular characterization and mapping of Fgf21 gene in a foodfish species Asian seabass. PLoS One 9:e90172
Wang X, Liu S, Jiang C, Geng X, Zhou T, Li N, Bao L, Li Y, Yao J, Yang Y (2017c) Multiple across-strain and within-strain QTLs suggest highly complex genetic architecture for hypoxia tolerance in channel catfish. Mol Gen Genomics 292:63–76
Xia JH, Lin G, He X, Liu P, Liu F, Sun F, Tu R, Yue GH (2013) Whole genome scanning and association mapping identified a significant association between growth and a SNP in the IFABP-a gene of the Asian seabass. BMC Genomics 14:295
Yue G, Wang L (2017) Current status of genome sequencing and its applications in aquaculture. Aquaculture 468:337–347
Yue GH (2014) Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish 15:376–396
Acknowledgements
This research was supported by the National Research Foundation, Prime Minister’s Office, Singapore, under its Competitive Research Program (CRP Award No. NRF-CRP7-2010-01) and TLL’s Innovation Support Fund (2016-54-0026).
Author information
Authors and Affiliations
Contributions
LW and GHY designed the experiments. LW, PL, ZYW, BY, and JW performed the virus challenge. LW, SH, and ZYW conducted the lab experiments. LW and BB analyzed the data. LW and GHY drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
The handling of animals in this study strictly followed the instructions of the Institutional Animal Care and Use Committee (IACUC) of Temasek Life Sciences Laboratory, Singapore (license no. TLL (F)-13-003).
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

{kind=link}
Cite this article
Wang, L., Bai, B., Huang, S. et al. QTL Mapping for Resistance to Iridovirus in Asian Seabass Using Genotyping-by-Sequencing. Mar Biotechnol 19, 517–527 (2017). https://doi.org/10.1007/s10126-017-9770-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-017-9770-8