
Abstract
Hizikia fusiforme, a brown seaweed, has been utilized as a health food and in traditional medicine. In this study, we investigated whether enzyme-modified H. fusiforme extracts (EH) have immunological effects compared with normal H. fusiforme extracts (NH). The effects of NH and EH on immune responses were investigated by assessing nitric oxide (NO) production, phagocytosis, and cytokine secretion in RAW 264.7 murine macrophages and mice. Also, fucosterol was evaluated to find the active component of NH and EH by addressing cytotoxicity test and NO production. Both of NH and EH significantly increased cell viability and NO synthesis. Tumor necrosis factor-α (TNF-α) expression was more induced by EH with LPS treatment. Phagocytic activity, as the primary function of macrophages, was markedly induced by EH treatment. Additionally, EH encouraged splenocyte proliferation and recovered the levels of cytokines IL-1β, IL-6, and TNF-α in mice. Finally, fucosterol increased NO production with no cytotoxicity, which means that fucosterol is an active component of EH. In conclusion, EH has the potential to modulate immune function and could offer positive therapeutic effect for immune system diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In order to efficiently overcome infection, immunomodulation requires the orchestration of many immune cells through signaling molecules in order to boost the immune system. Because the primary immune response initiates within macrophages, they are the first target of immunomodulators. The immune cells, i.e., macrophages, are present in almost all tissues and body cavities, including the peritoneal cavity. Macrophages which are monocyte-derived phagocytic cells play crucial roles in innate and adaptive immunities. The phagocytic function of macrophages can reflect immune function to some extent (Geissmann et al. 2010). During the course of an immune response, activated immune cells prevent pathogen invasion by secreting pro-inflammatory mediators such as nitric oxide (NO) and inflammatory cytokines including IL-1β, IL-6, and tumor necrosis factor-α (TNF-α) (Nathan 1987; Moncada and Higgs 1991). In particular, TNF-α is involved in systemic inflammation and is a member of a group of cytokines that stimulate the acute phase reaction (Riches et al. 1996). TNF-α is produced chiefly by activated macrophages, although it can also be produced by other cell types. NO, a product of macrophages activated by cytokines, has been recognized as one of the most versatile compounds in the immune system. Although NO has anti-cancer and anti-microbial effects, excessive secretion of NO can result in oxidative stress (Moncada and Higgs 1991). In the body, the spleen which is an important immune organ contains a relatively homogenous fraction of B and T lymphocytes, consisting of ∼60% B cells and 40% T cells. Splenocytes have different immune functions. Thus, an evaluation of the splenocyte proliferative effect provides an understanding of the influence on T and B cells (Cesta 2006).
In mammalian cells, the immune response is influenced by several essential nutrients that alter immune system function. During the last decade, an upsurge of information has surfaced concerning the immunological role of plant-derived materials in traditional medicine (Das et al. 2014). Brown seaweeds are traditionally consumed as food ingredients in East Asia. They contain large quantities of dietary fiber, minerals, vitamins, and polysaccharides (Rupérez and Saura-Calixto 2001; Bhaskar and Miyashita 2005; Mabeau and Fleurence 1993; Zhang et al. 2012); possess anti-cancer activities; and strengthen the immune response (Moussavou et al. 2014; Hwang et al. 2010). Hizikia fusiforme is a popular edible seaweed that is mainly composed of fucoses and sulfate bases (Patankar et al. 1993). Previous studies revealed that H. fusiforme modulates the immune system through several mechanisms, including induction of lymphocytes proliferation (Shan et al. 1999), cytokine synthesis (Yoon et al. 2011), and an increase of phagocytosis in macrophages (Choi et al. 2005). However, there are no reports regarding the ability of enzymatic preparations of H. fusiforme to modulate the immune response.
Among the components of H. fusiforme, fucosterol and fucoidan exert many biological activities such as anti-coagulant (Kuznetsova et al. 2003), anti-cancer (Zorofchian Moghadamtousi et al. 2014), anti-inflammatory (Park et al. 2011; Jung et al. 2013), anti-viral (Feldman et al. 1999), intestinal regulation (Iraha et al. 2013), and lipid-lowering effects (Huang et al. 2010). Recently, we investigated the protective effects of fucosterol from H. fusiforme against skin damage in UVB-irradiated human dermal fibroblasts (Hwang et al. 2014). Fucoidan is a heterogeneous assemblage with regard to monosaccharide composition, number of sulfate groups and acetyl groups, and molecular mass (Li et al. 2008). It has been reported that the biological activity of fucoidan depends on its structure and sulfate content (Jiao et al. 2011). Because enzymatic modification reduces the molecular weight of fucoidan and prevents the loss of sulfate groups, fucoidan is a useful functional food material. In this study, we performed enzyme modification of H. fusiforme in order to generate enzyme-modified H. fusiforme extracts (EH) with low-molecular weight fucoidan and fucosterol as active components. In this study, we investigated whether EH modulates the immune response by assessing splenocyte proliferation and cytokine production in in vitro and ex vivo models.
Materials and Methods
Preparation of EH
H. fusiforme was obtained from Wan-do Su-hyup (Wan-do, Korea) and extracted for 4 h in ethanol at a dried material to solvent ratio of 1:10 (w/v). Briefly, the crude enzyme was extracted from Aspergillus niger (KACC 40280), which was isolated from the Nuruk of makgeolli, a Korean traditional wine. A. niger was incubated on modified yeast mold agar (per liter distilled water 1.5 g beef extract, 3.0 g yeast extract, 2.5 g polypeptone, 0.05 g MgSO4, and 0.05 g CaCl2; pH 7.2) at 30 °C for 4 days. A. niger was cultured routinely on modified yeast mold broth at 30 °C. The sample was then centrifuged at 7000 rpm for 30 min. The crude enzyme was mixed with acetone at 4 °C for 4 h, and impurities were removed by precipitation in phosphate buffer. For purification, the active enzyme was loaded into an ion-exchange resin (QAEsephadex ® A50 and Sp-sephadex® C50, Sigma, St. Louis, USA) and eluted in NaCl buffer (Sigma). H. fusiforme extract was incubated with the enzyme at 60 °C for 24 h. After reaction, the enzyme was inactivated by incubation for 1 h at 100 °C. The EH were collected by filtration (Whatman no. 2 paper, USA), and the solvent was evaporated under reduced pressure and stored at −20 °C.
Analysis of EH
Fucoidan and fucosterol has been analyzed and previously characterized by our research group (Lee et al. 2015; Hwang et al. 2014). The molecular heterogeneity of purified fucoidan was studied by means of size exclusive HPLC using three tandem ultrahydrogel columns (1000, 500, and 250; Waters, USA). The analysis was performed with the following conditions: mobile phase 0.2 M KNO3, flow 0.9 mL/min, detection with RID, and attenuation of 16. Injection volume for samples with concentration 5–50 mg/mL. A Waters 600 controller equipped with a 996 PDA detector and a dual programmable pump (isocratic mode) operated with Emprover Pro® software was used for analysis. The tandem column system was calibrated using 5, 20, 100, and 500 kDa dextran sulfate (Sigma-Aldrich, USA).
For analysis of fucosterol from normal H. fusiforme extracts (NH) and EH, a fucosterol standard was purchased from Sigma-Aldrich (St. Louis, MO, USA). Analysis of the fucosterol was performed using a Waters LC/ESI-MS system equipped with a 2795 Alliance HT separation module, 2996 PDA detector, and Acquity SQD mass analyzer in positive ion mode. Chromatographic separation was carried out on a Waters C18 (0.46 × 25 cm) column. The mobile phase consisted of (A) water containing 0.1% formic acid and (B) acetonitrile containing 0.1% formic acid. Elution was performed as follows: 0–5 min, 80% B; 15 min, 100% B; 20 min, 80% B; and 25 min, 80% B. The flow rate was 1 mL/min and the injection volume was 10 μL. The LC/MS system employed MassLynx software (Waters, Milford, MA, USA). The following conditions were used: ion source temperature 150 °C, capillary voltage 3.5–4.5 kV, cone voltage 35–90 V (indicated for each experiment separately), dryer gas (N2) flow 500 L/h, and cone gas 50 L/h. Mass scanning ranged from 50 to 2000 kDa, and scanning speed was 0.2 s/scan.
Cell Culture
The murine macrophage cell line RAW 264.7 was obtained from KCLB (Seoul, Korea) and maintained in Dulbecco’s modified Eagle’s medium (PAA, Canada) containing 10% fetal bovine serum (PAA, Canada) and 1% penicillin-streptomycin (PAA, Canada) at 37 °C in a 5% CO2 incubator. RAW 264.7 cells were subcultured twice a week, and the medium was changed every 2 days. For experiments, samples were evaluated in the presence of various concentrations (1, 10, and 100 μg/mL) of NH and EH. The stock sample solution of NH and EH was prepared with DMSO (Sigma-Aldrich) as 10 mg/mL. Control cells were incubated for 24 h in the absence of samples.
MTT Assay
Cell viability was measured using a standard 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; thiazolyl blue) assay with slight modification (Lee et al. 2015). RAW 264.7 cells were seeded in 48-well plates at 5 × 104 cells/well for 24 h. Cell supernatants were removed and replaced with the appropriate concentrations of medium and samples. After incubation for 24 h, the cell supernatants were removed, MTT reagent (final concentration 0.1 mg/mL) was added, and the cells were incubated for 4 h. The cell supernatants were then removed, and DMSO was added to solubilize the insoluble formazan product generated by the mitochondrial activity of live cells. One hour later, the absorbance was measured with an enzyme-linked immunoassay (ELISA) microplate reader (Bio-Rad, Hercules, CA) at 570 nm. All experiments were performed in triplicate, and the relative cell viability (%) was expressed as a percentage relative to the untreated control cells.
Nitric Oxide Detection
NO production by RAW 264.7 cells was measured in aliquots of culture medium using the Griess reagent system (Promega, Madison, WI; Park et al. 2014). Cells were seeded in 24-well plates at 5 × 104 cells/well and incubated for 24 h. Samples were then added, and the incubation continued for an additional 24 h. Supernatant aliquots (50 μL) were transferred in triplicate into a 96-well plate, and 50 μL of sulfanilamide solution was added to each well. The plates were incubated for 10 min at room temperature, then N-(1-naphthyl)-N-1-ethylenediamine hydrochloride solution was added to the wells, and the plates were incubated for an additional 10 min at room temperature. The absorbance was then read at 540 nm using an ELISA microplate reader. Synthesized NO2 concentrations were determined by comparison to the nitrite standard reference curve.
Phagocytosis Assay
Phagocytosis assay was determined using commercially available ELISA kits (Cytoselect™ 96-well phagocytosis assay kit; Cell Biolabs, San Diego, CA, USA) according to the manufacturer’s instructions. Zymosan is a commonly used pathogen in phagocytosis assays. Cytochalasin D (2 μM) was used as inhibitor (blocks phagocytosis by interacting with actin microfilaments). The absorbance was then read at 405 nm using an ELISA microplate reader. The phagocytosis response to NH and EH was expressed as follows
Measurement of IL-1β, IL-6, and TNF-α
To assess the production of pro-inflammatory cytokines by RAW 264.7 cells and mouse peritoneal cells, we used commercially ELISA kits. Cells were seeded in a 24-well plate, and various concentrations of NH and EH were added to the wells. The cells were then incubated for an additional 24 h, after which the cell medium was collected from each well. The concentrations of IL-1β, IL-6, and TNF-α were analyzed from cell medium using commercially available ELISA kits (R&D Systems, Inc., Minneapolis, MN, USA) in accordance with the manufacturers’ instructions. Each sample was analyzed in triplicate. The absorbance was measured at 450 nm using an ELISA microplate reader. The production of pro-inflammatory cytokine was determined by comparison to the standard reference curve.
Treatment and Laboratory Animals
Seven-week-old male C57BL/6 mice weighing 24–26 g (BDL, Chungbuk, Korea) were used and were housed in the laboratory at 22 ± 2 °C and 40–60% humidity with a 12 h light/12 h photoperiod. The mice had free access to solid feed and water and were allowed to acclimatize for 1 week before the experiment. After 1 week acclimation, samples were dissolved in distilled water and administered orally for 3 weeks. Total 35 animals were divided into five groups of different NH and EH doses. The groups with each seven mice were further divided into subgroups (NC: normal control group, NH: orally administered H. fusiforme extract at 200 mg/kg body weight [bw], and EH: orally administered enzyme-treated low-molecular weight fucoidan from H. fusiforme at 50, 100, and 200 mg/kg bw). NH and EH were provided on alternate days throughout the experimental period. The experimental protocol [KHUASP(SE)-15-027] was approved by the Institutional Animal Care and Use Committee of Kyung Hee University.
Measurement of Splenocyte Proliferation
Splenocytes were separated using the method of Mishell (Mishell and Shiigi 1980). To measure splenocyte proliferation, splenocyte suspensions for each group were diluted and plated on 96-well plates (Corning, Corning, NY, USA). Con A (5 μg/mL) and lipopolysaccharide (LPS) (15 μg/mL; Sigma Co., St. Louis, MO, USA) were used as mitogens. Optical density was measured using an ELISA reader at 540 nm for the MTT assay (Małaczewska 2014). The results of the proliferation assay (%) were calculated by dividing the mean O.D. of stimulated cultures by the O.D. of the non-stimulated (control) cultures.
Isolation of Peritoneal Macrophages
After anesthetization using ether, the animals were exsanguinated, and the peritoneal cavities were washed with 5 mL of sterile FBS-free RPMI medium (10%, v/v), penicillin (100 units/mL), and streptomycin in an aseptic environment. Resulting cell suspensions were centrifuged for 10 min at 1000 RPM and 4 °C and then re-suspended in 10 mL of sterile RPMI medium without glutamine. Macrophages were obtained by incubating 1 × 109 cells per dish in 24-well polystyrene culture plates for 2 h. Non-adherent cells were removed by three vigorous washes with RPMI medium without glutamine. The entire procedure was executed under aseptic conditions, and all materials were previously sterilized.
Statistical Analysis
Data are expressed as mean value ± standard deviation (SD). Based on analysis of variance (ANOVA), statistically significant differences between groups (P < .05) were identified. These results were determined using the PRISM Statistical Analysis System. The statistical significance of differences between mean values was tested using the independent two-tailed Student’s t test in Sigma Plot 2001 software. Statistical significance was set at p < 0.05.
Results
Analysis of Fucoidan and Fucosterol from NH and EH
To confirm fucoidan and fucosterol from NH and EH, we used HPLC. As shown in Fig. 1a, molecular size of fucoidan from NH and EH were analyzed using size exclusion HPLC. The β-glycosidase from Aspergillus sp. exploits the hydrolytic pathways. The high molecular weight of fucoidan (500 kDa) transformed into low molecular weight of fucoidan (5 and 2–3 kDa). Because these β-d-glycosidases were able to simultaneously hydrolyze, they produced the low molecular weight of fucoidan in EH. HPLC was used to confirm that fucosterol was extracted from NH and EH. As shown in Fig. 1b, the retention time for the fucosterol standard was 15.48 min. Fucosterol peaks from NH and EH were identified by comparing retention times with those of the corresponding standards. The total fucosterol concentration in NH and EH was calculated as 0.249 and 1.067 mg/g (Fig. 1b).


A size exclusion HPLC results of molecular size of fucoidan from NH and EH (a) and HPLC results of fucosterol from NH and EH (b)
Effect of EH on Cell Viability in RAW 264.7 Cells
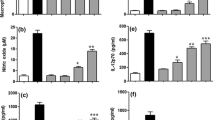
To determine the optimal concentrations of NH and EH for all experiments, the effects of NH and EH on the viability of RAW 264.7 cells were primarily investigated using MTT assay. As demonstrated in Fig. 2a, treatment with NH and EH at all concentrations between 1 and 100 μg/mL did not significantly reduce cell viability. At high concentrations of NH and EH (100 μg/mL), cell viability was rather increased by 128.5 ± 5.3 and 150.3 ± 2.3%, respectively, and which values were statistically significant. For this reason, EH were used at this concentration in all subsequent experiments.

Cell viability (a), NO production (b), and TNF-α expression (c) in NH- or EH-treated RAW 264.7 cells. The cells were treated for 24 h. LPS (1 μg/mL) was use as a positive control. d Measurement of phagocytosis in NH- or EH-treated RAW 264.7 cells that were treated for 24 h. Zymosan was used a pathogen. Cytochalasin D (2 μM) was used as inhibitor. The absorbances were then read at 570, 540, 450, and 405 nm, respectively, using a microplate reader. Data are presented as mean ± S.D. of three independent experiments performed in triplicate. Number sign and asterisk indicate significant differences (p < 0.05) between the LPS (−) control (normal) and LPS (+) control, respectively. ###p < 0.001 versus the normal, *p < 0.05, **p < 0.01, and ***p < 0.001 versus LPS-induced control
Effect of EH on NO Production in RAW 264.7 Cells
To assess the effects of NH and EH on NO production in RAW 264.7 cells, we measured the nitrite concentration in culture medium using Griess reagent (Fig. 2b). As expected, LPS treatment greatly stimulated NO production by 36.2 ± 5.1% compared to the untreated cells. As shown in Fig. 2b, treatment with NH and EH at 100 μg/mL resulted in NO production increases of 24.3 ± 1.9 and 29.1 ± 2.5%, respectively, which were statistically significant. These results indicate that NO production is more potent with EH treatment than with NH treatment.
Effect of EH on TNF-α Production in RAW 264.7 Cells
As the regulator of immune cells, TNF-α induces other cytokines and stimulates macrophage phagocytosis. As shown in Fig. 2c, TNF-α production was increased by LPS treatment (up to 145%) compared to that in non-LPS-treated cells. NH in LPS-treated cells increased TNF-α production by 159% at 100 μg/mL compared to that in NH-treated cells without LPS. Also, EH in LPS-treated cells significantly increased TNF-α production by 180% at 100 μg/mL compared to that of EH-treated cells without LPS. These results support the conclusion that EH has a stronger immune-stimulating effect than NH.
Effect of EH on Phagocytosis in RAW 264.7 Cells
In the present study, we examined the effects of NH and EH on phagocytosis in macrophages. When RAW 264.7 cells were treated with NH (100 μg/mL) and EH (1–100 μg/mL) for 24 h, phagocytic activity was significantly increased (Fig. 2d). At the highest concentration, phagocytic activity of EH-treated cells (up to 128.3 ± 2.3% at 100 μg/mL) was greater than that of NH-treated cells (up to 118.4 ± 1.6%).
Effect of orally Administered EH on Splenocyte Proliferation
As shown in Fig. 3a, splenocyte proliferation was determined after day 21 of NH or EH oral administration. Splenocyte proliferation was induced by Con A activation, which selectively induces T cell proliferation. The EH 200 mg/kg group showed higher splenocyte proliferation (up to 38%) than that of the Con A-only control group after day 21. Treatment with LPS selectively induces B cell proliferation and then activates splenocyte proliferation. Although EH stimulated splenocyte proliferation in the LPS-activated condition compared to no treatment in the EH control, the difference was not significant. After treatment with Con A and LPS, the EH group showed the highest level of splenocyte proliferation.

a Proliferation of splenocytes in mouse orally administered NH or EH treated for 21 days. Con A (5 μg/mL) and LPS (15 μg/mL) selectively induced T cell and B cell proliferation, respectively. The data represents the mean ± SD of quadruplicate experiments. Number sign and asterisk indicate significant differences (p < 0.05) between the without mitogen (normal) and only Con A-treated control, respectively. ##p < 0.01 versus the normal, **p < 0.01 versus only Con A-treated control. TNF-α production (b), IL-1β secretion (c), and IL-6 expression (d) in mouse orally administered NH or EH treated for 21 days. The data represents the mean ± SD of quadruplicate experiments. Number sign and asterisk indicate significant differences (p < 0.05) between the LPS (−) control (normal) and the LPS (+) control, respectively. #p < 0.05 and ##p < 0.01 versus the normal, *p < 0.05 and **p < 0.01 versus LPS-induced control
Effects of Orally Administered EH on Cytokine Expression in Mouse Peritoneal Macrophages
EH is well established as an immunomodulatory agent. Hence, we studied the effect of EH treatment on immune response modulation via cytokine (IL-1β, IL-6, and TNF-α) expression in murine peritoneal macrophages. We observed that LPS increases in the release of IL-1β, IL-6, and TNF-α by 138, 171, and 181%, respectively, in comparison with the non-LPS-treated group in peritoneal macrophages. At the LPS-treated condition, EH (100 μg/mL) increased IL-1β, IL-6, and TNF-α production by 112, 117, and 129%, respectively, compared to that in only LPS treatment. Especially, EH 200 μg/mL group showed high level of statistical significance in IL-6 production. The significant increases in IL-1β, IL-6, and TNF-α release might play a pivotal role in triggering the signal for enhanced immune response. These upregulated secretions were more prominent in EH than in NH samples (Fig. 3b–d).
Effect of Fucosterol on Cell Viability and NO Production in RAW 264.7 Cells
To evaluate the effects of fucosterol, an active component of EH, we measured cell viability and NO production in RAW 264.7 cells. No cytotoxicity was observed at any fucosterol concentration (Fig. 4a). At a high fucosterol concentration (50 μg/mL), cell viability was increased by 112%. The NO production was the highest in the LPS-treated RAW 264.7 cells (up to 313%). Fucosterol also stimulated NO secretion by 132% at 25 μg/mL and 135% at 50 μg/mL (Fig. 4b).

Cell viability (a) and NO production (b) in fucosterol-treated RAW 264.7 cells. The cells were treated for 24 h. LPS (1 μg/mL) was use as a positive control. Data are presented as mean ± S.D. of three independent experiments performed in triplicate. Number sign and asterisk indicate significant differences (p < 0.05) between the LPS (−) control (normal) and LPS (+) control, respectively. ###p < 0.001 versus the normal, *p < 0.05 and **p < 0.01 versus LPS-induced control
Discussion
Since natural products have a relatively low potential toxicity, they are useful in clinical application. It is an increasing trend to use natural products as candidate immunomodulatory agents. However, biological activities and application of marine algae have not been sufficiently investigated. Recently, Mayer et al. reported that several marine compounds have regulatory effects on the immune system (Mayer et al. 2013). Reports about H. fusiforme include that of Jeong et al., who showed the immune-modulating activities of polysaccharides extracted from H. fusiforme (Jeong et al. 2015). However, no immune activities from the extracts of enzyme-modified H. fusiforme have been reported nor have their biological mechanisms been elucidated. In this study, we investigated the immunomodulatory effects of EH on LPS-induced murine macrophages in vitro and orally administered mice ex vivo.
To modify H. fusiforme, crude extracts were treated with an enzyme and low-molecular weight fucoidan and fucosterol were analyzed from EH. The activity of fucoidan on immunomodulation has been previously reported. Nakamura et al. reported that fucoidan increased the level of nitric oxide (NO) production in macrophages in relation to p38 kinase-dependent NF-κB activation (Nakamura et al. 2006), and Maruyama H et al. studied the oral administration of fucoidan to T cell receptor transgenic (DO11.10-Tg) mice with increased anti-tumor activities due to enhanced T helper type 1 (Th1) immune responses and resultant T cell-mediated NK cell activation (Maruyama et al. 2006). However, fucoidan is chemically unstable, and its bioactivity is highly dependent on the number of sulfate groups. Therefore, to overcome these weaknesses, we attempted to transform NH into EH via enzymatic modification that prevents loss of sulfate groups and results in a lower molecular weight. Previously, our group demonstrated that the anti-aging effect of enzyme-treated ginseng extract was better than that of pure ginseng extract (Hwang et al. 2013). For this reason, we utilized enzymatic modification in the present study. As expected, the results showed that EH which has the low molecular weight of fucoidan stimulated a stronger immune response than did pure NH.
Activated macrophages are important for the initiation and perpetuation of specific immune responses, both by serving as antigen-presenting cells and by generating cytokines that promote proliferation, differentiation, and function of T lymphocytes, as well as other diverse immune effector cells (Mosser and Edwards 2008). Pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α are stimulated by LPS, which is present in the outer cell membranes of gram-negative bacteria, in macrophages and monocytes (Bruunsgaard et al. 1999). The primary role of TNF-α is the regulation of immune cells. TNF-α induces other cytokines such as IL-1β (Melchiorri et al. 1998), IL-6 (Turner et al. 2007), and IL-8 (Zhao et al. 2005). In this study, EH induced the secretion of TNF-α in LPS-treated murine macrophage cells. In addition, all levels of IL-1β, IL-6, and TNF-α increased upon EH treatment in peritoneal macrophages. These changes were significant when stimulated by LPS, whereas they were not observed without LPS. This finding suggests that the stimulatory effects of pro-inflammatory cytokines in EH have synergistic activity with LPS treatment in macrophage.
Phagocytosis is the primary function of macrophages and enhances a diverse range of anti-microbial and cytotoxic responses including generation of respiratory burst, secretion of immune mediators, and antigen presentation (Greenberg and Grinstein 2002; Aderem and Underhill 1999). As a part of the immune response, NO is generated by phagocytes (monocytes, macrophages, and neutrophils) (Aderem 2003). To regulate immune responses, NO triggers the elimination of pathogens on the one hand and modulates immunosuppression during tissue-restoration and wound healing processes on the other. Interestingly, fluctuations in the levels of NO orchestrate other phases of the immune response. NO activates specific signal transduction pathways in tumor cells, endothelial cells, and monocytes in a concentration-dependent manner (Wink et al. 2011), whereas excessive production of NO during inflammation may cause tissue injury, nerve damage, and edema resulting from increased blood vessel permeability (Garlanda et al. 2007; Kaur et al. 2007). In addition, increased TNF-α stimulates macrophage phagocytosis (Hess et al. 2009).
We confirmed that fucosterol, as an active component of EH, increased NO production with no cytotoxicity. When NH were transformed to EH through enzymatic treatment, fucosterol content was significantly increased. Based on these observations, EH has stronger effects on phagocytic activity and NO production than does NH, although the immunostimulatory effects of NH are stronger after enzymatic modification. Based on these immunomodulatory effects of EH in murine macrophages, further ex vivo studies were performed to examine whether EH also stimulates immune-related cytokine and mouse splenocyte proliferation in mice treated with oral EH. The spleen is a major organ, and the size or proliferation of splenocytes, increased by Con A activation via induction of T cell proliferation, is an indicator of immune function. Splenocytes participate principally in innate (monocytes and NK cells) and acquired (T and B cells) immune defenses, and previous studies have demonstrated that the effects of natural food and herbal products on host defense against microbial pathogens and tumors were directly correlated with their ability to stimulate lymphocyte proliferation (Ho et al. 2004; Lee et al. 2005). Splenocyte proliferation stimulated by medicinal fruits and vegetables has been attributed to their high concentration of phenolic compounds (Lin and Tang 2007). Previous studies revealed that H. fusiforme modulates the immune system through several mechanisms; however, there are no reports regarding the ability of EH to modulate the immune response. We confirmed that the group treated with EH showed greater splenocyte proliferation than that in the only Con A-treated control group, even compared with NH treated group.
In summary, the results of this study verified that EH shows greater immune stimulation than pure NH. More specifically, EH increased secretion of TNF-α, production of NO, and phagocytosis activity in murine macrophage cells. EH contains a higher level of the active component fucosterol than does NH. Moreover, due to the modification of high-molecular weight fucoidan in NH to low-molecular weight fucoidan in EH via enzyme treatment, the stimulatory effect of EH was higher than that of NH. However, the effective EH dose may depend on physical parameters. Therefore, further studies are needed to explore EH in relation to physical parameters through diverse molecular weights of fucoidan, as well as to investigate the action mechanisms of EH. To the best of our knowledge, the immunostimulatory effects of EH rich in the active component fucosterol and low-molecular weight fucoidan were established here for the first time. We expect our findings to be used as a basis for developing functional food products that contain EH.
References
Aderem A (2003) Phagocytosis and the inflammatory response. J Infect Dis 187:S340–S345
Aderem A, Underhill DM (1999) Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 17:593
Bhaskar N, Miyashita K (2005) Lipid composition of Padina tetratomatica (Dictyotales, Pheophyta), a brown seaweed of the west coast of India. Indian J Fish 52:263–268
Bruunsgaard H, Pedersen AN, Schroll M, Skinhøj P, Pedersen BK (1999) Impaired production of proinflammatory cytokines in response to lipopolysaccharide (LPS) stimulation in elderly humans. Clin Exp Immunol 118(2):235–241
Cesta MF (2006) Normal structure, function, and histology of the spleen. Toxicol Pathol 34(5):455–465
Choi EM, Kim AJ, Kim Y, Hwang JK (2005) Immunomodulating activity of arabinogalactan and fucoidan in vitro. J Med Food 8:446–453
Das S, Bordoloi R, Newar N (2014) A review on immune modulatory effect of some traditional medicinal herbs. J Pharm Chem Biol Sci 2(1):33–42
Feldman SC, Reynaldi S, Stortz CA, Cerezo AS, Damont EB (1999) Antiviral properties of fucoidan fractions from Leathesia difformis. Phytomedicine 6(5):335–340
Garlanda C, Di Liberto D, Vecchi A, La Manna MP, Buracchi C, Caccamo N, Salerno A, Dieli F, Mantovani A (2007) Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J Immunol 179(5):3119–3125
Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K (2010) Development of monocytes, macrophages, and dendritic cells. Science 327:656–661
Greenberg S, Grinstein S (2002) Phagocytosis and innate immunity. Curr Opin Immunol 14:136
Hess DJ, Henry-Stanley MJ, Bendel CM, Zhang B, Johnson MA, Wells CL (2009) Escherichia coli and TNF-alpha modulate macrophage phagocytosis of Candida glabrata. J Surg Res 155:217–224
Ho CY, Lau CB, Kim CF, Leung KN, Fung KP, Tse TF (2004) Differential effect of Coriolus versicolor (Yunzhi) extract on cytokine production by murine lymphocytes in vitro. Int Immunopharmacol 4:1549–1557
Huang L, Wen K, Gao X, Liu Y (2010) Hypolipidemic effect of fucoidan from Laminaria japonica in hyperlipidemic rats. Pharm Biol 48:422–426
Hwang PA, Wu CH, Gau SY, Chien SY, Hwang DF (2010) Antioxidant and immune-stimulating activities of hot-water extract from seaweed Sargassum hemiphyllum. J Mar Sci Tech 18:41–46
Hwang E, Sun ZW, Lee TH, Shin HS, Park SY, Lee DG, Cho BG, Sohn H, Kwon OW, Kim SY, Yi TH (2013) Enzyme-processed Korean Red Ginseng extracts protects against skin damage induced by UVB irradiation in hairless mice. J Ginseng Res 37(4):425–434
Hwang E, Park SY, Sun ZW, Shin HS, Lee DG, Yi TH (2014) The protective effects of fucosterol against skin damage in UVB-irradiated human dermal fibroblasts. Mar Biotech 16(3):361–370
Iraha A, Chinen H, Hokama A, Yonashiro T, Kinjo T, Kishimoto K, Nakamoto M, Hirata T, Kinjo N, Higa F, Tateyama M, Kinjo F, Fujita J (2013) Fucoidan enhances intestinal barrier function by upregulating the expression of claudin-1. World J Gastroenterol 19(33):5500–5507
Jeong SC, Jeong YT, Lee SM, Kim JH (2015) Immune-modulating activities of polysaccharides extracted from brown algae Hizikia fusiforme. Biosci Biotechnol Biochem 79(8):1362–1365
Jiao G, Yu G, Zhang J, Ewart HS (2011) Chemical structures and bioactivities of sulfated polysaccharides from marine algae. Mar Drugs 9(2):196–223
Jung HA, Jin SE, Ahn BR, Lee CM, Choi JS (2013) Anti-inflammatory activity of edible brown alga Eisenia bicyclis and its constituents fucosterol and phlorotannins in LPS-stimulated RAW264.7 macrophages. Food Chem Toxicol 59:199–206
Kaur C, Sivakumar V, Lu J, Ling EA (2007) Increased vascular permeability and nitric oxide production in response to hypoxia in the pineal gland. J Pineal Res 42(4):338–349
Kuznetsova TA, Besednova NN, Mamaev AN, Momot AP, Shevchenko NM, Zvyagintseva TN (2003) Anticoagulant activity of fucoidan from brown algae Fucus evanescens of the Okhotsk Sea. Bull Exp Biol Med 5:471–473
Lee SH, Park JB, Park HJ, Park YJ, Sin JI (2005) Biological properties of different types and parts of the dandelions: comparisons of anti-oxidative, immune cell proliferative and tumor cell growth inhibitory activities. Prev Nutr Food Sci 10:172–178
Lee DG, Park SY, Chung WS, Park JH, Hwang E, Mavlonov GT, Kim IH, Kim KY, Yi TH (2015) Fucoidan prevents the progression of osteoarthritis in rats. J Med Food 18(9):1032–1041
Li B, Lu F, Wei X, Zhao R (2008) Fucoidan: structure and bioactivity. Molecules 13:1671–1695
Lin JY, Tang CY (2007) Determination of total phenolic and flavonoid contents in selected fruits and vegetables, as well as their stimulatory effects on mouse splenocyte proliferation. Food Chem 101:140–147
Mabeau S, Fleurence J (1993) Seaweed in food products: biochemical and nutritional aspects. Trends Food Sci Technol 4:103–107
Małaczewska J (2014) The splenocyte proliferative response and cytokine secretion in mice after 28-day oral administration of silver nanocolloid. Pol J Vet Sci 17(1):27–35
Maruyama H, Tamauchi H, Iizuka M, Nakano T (2006) The role of NK cells in antitumor activity of dietary fucoidan from Undaria pinnatifida sporophylls (Mekabu). Planta Med 72:1415–1417
Mayer AM, Rodríguez AD, Taglialatela-Scafati O, Fusetani N (2013) Marine pharmacology in 2009–2011: marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar Drugs 11(7):2510–2573
Melchiorri C, Meliconi R, Frizziero L, Silvestri T, Pulsatelli L, Mazzetti I, Borzì RM, Uguccioni M, Facchini A (1998) Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum 41(12):2165–2174
Mishell BB, Shiigi SM (1980) Selected methods in cellular immunology, 1st edn. WH Freeman and Co., San Francisco, pp 4–27
Moncada S, Higgs EA (1991) Endogenous nitric oxide: physiology, pathology and clinical relevance. Eur J Clin Investig 21(4):361–374
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8(12):958–969
Moussavou G, Kwak DH, Obiang-Obonou BW, Maranguy CA, Dinzouna-Boutamba SD, Lee DH, Pissibanganga OG, Ko K, Seo JI, Choo YK (2014) Anticancer effects of different seaweeds on human colon and breast cancers. Mar Drugs 12(9):4898–4911
Nakamura T, Suzuki H, Wada Y, Kodama T, Doi T (2006) Fucoidan induces nitric oxide production via p38 mitogen-activated protein kinase and NF-kappaB-dependent signaling pathways through macrophage scavenger receptors. Biochem Biophys Res Commun 343(1):286–294
Nathan CF (1987) Secretory products of macrophages. J Clin Invest 79:319–326
Park HY, Han MH, Park C, Jin CY, Kim GY, Choi IW, Kim ND, Nam TJ, Kwon TK, Choi YH (2011) Anti-inflammatory effects of fucoidan through inhibition of NF-κB, MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia cells. Food Chem Toxicol 49(8):1745–1752
Park SY, Kim HB, Kim JH, Lee JM, Kim SR, Shin HS, Yi TH (2014) Immunostimulatory effect of fermented red ginseng in the mouse model. Prev Nutr Food Sci 19(1):10–18
Patankar MS, Oehninger S, Barnett T, Williams RL, Clark GF (1993) A revised structure for fucoidan may explain some of its biological activities. J Biol Chem 268:21770–21776
Riches DW, Chan ED, Winston BW (1996) TNF-alpha-induced regulation and signalling in macrophages. Immunobiology 195(4–5):477–490
Rupérez P, Saura-Calixto F (2001) Dietary fiber and physicochemical properties of edible Spanish seaweeds. Eur Food Res Technol 212:349–354
Shan BE, Yoshida Y, Kuroda E, Yamashita U (1999) Immunomodulating activity of seaweed extract on human lymphocytes in vitro. Int J Immunopharmacol 21(1):59–70
Turner NA, Mughal RS, Warburton P, O’Regan DJ, Ball SG, Porter KE (2007) Mechanism of TNFα-induced IL-1α, IL-1β and IL-6 expression in human cardiac fibroblasts: effects of statins and thiazolidinediones. Cardiovasc Res 76:81–90
Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA (2011) Nitric oxide and redox mechanisms in the immune response. J Leukoc Biol 89(6):873–891
Yoon YD, Lee ES, Park JP, Kim MR, Lee JW, Kim TH, Na MK, Kim JH (2011) Immunostimulatory effect by aqueous extract of Hizikia fusiforme in RAW 264.7 macrophage and whole spleen cells. Biotech Bioprocess Eng 16:1099–1105
Zhang CY, Wu WH, Wang J, Lan MB (2012) Antioxidant properties of polysaccharide from the brown seaweed Sargassum graminifolium (Turn.), and its effects on calcium oxalate crystallization. Mar Drugs 10(1):119–130
Zhao RZ, Chen X, Yao Q, Chen C (2005) TNF-alpha induces interleukin-8 and endothelin-1 expression in human endothelial cells with different redox pathways. Biochem Biophys Res Commun 327:985–992
Zorofchian Moghadamtousi S, Karimian H, Khanabdali R, Razavi M, Firoozinia M, Zandi K, Abdul Kadir H (2014) Anticancer and antitumor potential of fucoidan and fucoxanthin, two main metabolites isolated from brown algae. Sci World J 2014:1–10
Acknowledgements
The authors would like to thank YTH LifeScience LTD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The experimental protocol [KHUASP(SE)-15-027] was approved by the Institutional Animal Care and Use Committee of Kyung Hee University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Sang-Yong Park and Eunson Hwang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Park, SY., Hwang, E., Shin, YK. et al. Immunostimulatory Effect of Enzyme-Modified Hizikia fusiformein a Mouse Model In Vitro and Ex Vivo. Mar Biotechnol 19, 65–75 (2017). https://doi.org/10.1007/s10126-017-9727-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-017-9727-y