
Abstract
Sex dimorphic growth pattern has significant theory and application implications in fish. Recently, a Y- and X-specific allele marker-assisted sex control technique has been developed for mass production of all-male population in yellow catfish (Pelteobagrus fulvidraco), but the genetic information for sex determination and sex control breeding has remained unclear. Here, we attempted to provide the first insight into a comprehensive transcriptome covering multiple tissues from XX females, XY males, and YY super-males of yellow catfish by using 454 GS-FLX platform, for a better assembly and gene coverage. A total of 1,202,933 high quality reads (about 540 Mbp) were obtained and assembled into 28,297 contigs and 141,951 singletons. BLASTX searches against the NCBI non-redundant protein database (nr) led a total of 52,564 unique sequences including 18,748 contigs and 33,816 singletons to match 25,669 known or predicted unique proteins. All of them with annotated function were categorized by gene ontology (GO) analysis, and 712 were assigned to reproduction and reproductive process. Some potential genes relevant to reproductive system including steroid hormone biosynthesis and GnRH (gonadotropin-releasing hormone) signaling pathway were further identified by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis; and at least 21 sex determination and differentiation-related genes, such as Dmrt1, Sox9a/b, Cyp19b, WT1, and AMH were identified and characterized. Additionally, a total of 82,794 simple sequence repeats (SSRs), 26,450 single nucleotide polymorphisms (SNPs), and 4,145 insertions and deletions (INDELs) were revealed from the transcriptome data. Therefore, the current transcriptome resources highlight further studies on sex-control breeding in yellow catfish and will benefit future studies on reproduction and sex determination in teleost fish.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sex dimorphic growth pattern has been found in many species of teleost fish (Gui 2007; Mei and Gui 2014), and the significant growth difference between female and male and its application implications have been noted in some farmed fish (Taranger et al. 2010; Gui and Zhou 2010; Gui and Zhu 2012; Kobayashi et al. 2013; Liu et al. 2013a). Yellow catfish (Pelteobagrus fulvidraco), one of the important freshwater fish species in Asia, has been widely cultured for its delicious flesh and high nutritive value (Huang et al. 1999). According to a previous survey on yellow catfish, males exhibited much faster growth rate than female siblings under the same culturing condition (Liu et al. 2007), and this male priority phenomenon was also observed in many other fish species, such as Nile tilapia (Oreochromis niloticus) (Lee et al. 2011), African catfish (Clarias gariepinus) (Henken et al. 1987), and European catfish (Silurus glanis) (Haffray et al. 1998). Moreover, YY super-male and all-male techniques had been established by hormonal-induced sex reversal as well as Y- and X-specific allele markers to improve the culture of tilapia and yellow catfish (Sarder et al. 1999; Wang et al. 2009; Dan et al. 2013; Liu et al. 2013b). Especially in yellow catfish, an all-male monosex population, nominated as “yellow catfish all-male No. 1”, had been created by crossing YY super-male and XX female, and widely cultured for commercial purpose throughout China (Wang et al. 2009; Dan et al. 2013; Liu et al. 2013b). Significantly, the controllable XY all-males, especially the YY super-males, give us infrequent genetic resources to study candidate genes and possible pathways responsible for sex determination and differentiation (Xia et al. 2007; Huang et al. 2009; Zhou and Gui 2010; Gui and Zhu 2012; Xu et al. 2013; Mei et al. 2013), since most of the genetic information has remained unclear in this fish species. Moreover, there is no report about genetic selection breeding of this species until now, because of the lack of genomic data.
Although sex is determined by sex chromosome-linked genes Sry and Dmrt1 in mammals and birds (Sinclair et al. 1990; Smith et al. 2009), sex determination in fish is a plastic process that is controlled by both genetic and environmental factors (Baroiller et al. 2009). Interestingly, sex-determining genes were identified to be diverse in rainbow trout (Oncorhynchus mykiss), fugu (Takifugu rubripes), Patagonian pejerrey (Odontesthes hatcheri), and medaka (Oryzias latipes), and all of these genes are male-specific and located in the sex chromosome or sex-determining locus (Matsuda et al. 2002; Hattori et al. 2012; Kamiya et al. 2012; Yano et al. 2012). This variety of sex-determining genes among fish species suggests that plentiful of genetic resources and sex-related genes should be collected and characterized to elucidate sex determination mechanism in fish, especially in yellow catfish that has no report for genes involved in sex determination and differentiation. In response to these challenges, we here used high-throughput 454 pyrosequencing technology to explore the transcriptome of yellow catfish, because of its potential advantages of long sequencing reads and accuracy (Salem et al. 2010). From the assembled sequences, we identified many functional genes and their involved pathways that are related to sex determination and differentiation. In addition, we discovered a number of SSRs and SNPs that will greatly benefit the selective breeding in this species, which has so far been hampered by a lack of genomic data.
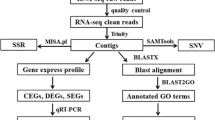
Methods
Samples Collection and Preparation
Fourteen 4-year-old yellow catfish individuals (six XX females, six XY males, and two YY super-males) were collected from our breeding center at Jingzhou, Hubei province, China. And their sex was confirmed by histological analysis and sex-linked makers as described previously (Dan et al. 2013). Experimental protocols used here were approved by the institution animal care and use committee of Huazhong Agricultural University. A total of nine major tissues, including three gonadal tissues (XX ovary, XY testis, YY testis) and six other tissues (liver, kidney, muscle, brain, spleen, and heart) were taken from each individuals of above. The brain tissues contained hypothalamus and pituitary. Finally, we combined the same type tissue into one tissue group, and each of these nine tissue groups has equal weight.
RNA Extraction, cDNA Library Construction, and 454 Sequencing
Total RNA was extracted from each of the nine tissue groups with TRIzol Reagent (Invitrogen, USA) according to the manufacturer’s instructions. After treating by Turbo Rnase-free DNase (Ambion), the extracted RNA samples were purified with RNeasy Mini Kit (QIAGEN, USA) following the manufacturer’s instruction. All the RNA samples were standardized to 200 ng/μL. RNA quality and quantity were analyzed using gel electrophoresis and NanoDrop (Thermo Scientific, USA). Then, we mixed equal quantity of total RNA from each tissue group into one RNA pool, which was used for complementary DNA (cDNA) library construction with SMART cDNA synthesis kit (Clontech Laboratories, USA). The 454 sequencing library was synthesized with the 454 GS FLX Titanium General Library Preparation Kit according to the manufacturer’s manual as described (Chen et al. 2013b), and performed on a single plate with Roche 454 GS FLX Titanium genomic sequencer at Shanghai OE Biotech Company.
Sequence Assembly and Annotation
For all raw reads, low-quality bases and the sequence of adapters were removed by LUCY and Seq-Clean software as previously reported (Liao et al. 2013). The reads were assembled using the Newbler software package (Roche). The raw sequencing data has been deposited to the NCBI archive database (accession number, SRP032172). Gene annotation was performed by local BLASTX searches against the NCBI non-redundant (nr), STRING, and GENE databases with a significance threshold of E ≤ 1e-5. Gene names were assigned to each sequence based on the highest alignment score among BLAST matches. To explore gene regulatory network in yellow catfish, the Gene Ontology (GO) database, Cluster of Orthologous Groups (COG) database, and the involvement of Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathway database were carried out for each annotated unique sequence.
Real-Time PCR Analyses
cDNA was synthesized using GoScript™ Reverse Transcription System (Promega) with RNA extracted from immature XX ovary, XY, and YY testis of 1-year-old yellow catfish. Real-time PCR was performed with IQ SYBR Green Supermix (Bio-Rad Laboratories) on the CFX96™ real-time system (Bio-Rad Laboratories) as described (Xiao et al. 2014; Li et al. 2014a). According to the sequences in our transcriptome, primers were designed using Primer Premier 5.0 software (Table S4). After incubation 95 °C for 5 min, the cycling protocol was followed by 39 cycles of 95 °C for 5 s, 55 °C for 15 s, 72 °C for 30s. After amplification, a melting curve was performed according to the manufacturer recommendations to check the amplification specificity as described (Li et al. 2014b). The specificity of amplification was also confirmed by agarose gel electrophoresis. Relative expression was normalized to β-actin gene.
SSRs, SNPs, and INDELs Marker Characterization
As previously described, SSRs were identified from the unique sequences using MISA (http://pgrc.ipk-gatersleben.de/misa/), with the parameters set to ≥10 repeat units for mononucleotide SSRs, ≥6 repeat units for dinucleotide, and ≥5 repeat units for trinucleotide, tetranucleotide pentanucleotide, and hexanucleotide SSRs (Liao et al. 2013). Two SSRs separated by >100 bp were considered to be two SSRs, and ≤100 bp as part of a compound SSR, respectively. We run bowtie2 (bowtie-bio.sourceforge.net/bowtie2/) and used samtools 0.1.18 (samtools.sourceforge.net/) to find SNPs and INDELs in a Bowtie output file.
Results and Discussion
Transcriptome Sequencing and Assembly
In order to comprehensively identify genes for functional genetic studies, we synthesized a pooled cDNA library from ovary, testis, liver, kidney, muscle, brain, spleen, and heart of the experimental yellow catfish, which was run on a 454 GS-FLX Titanium platform. After eliminating primer and adapter sequences, this sequencing generated 1,202,933 high-quality raw reads, with an average sequence length of 449.5 base pairs, and a total of 540 Mbp sequence data. A summary of the 454 sequence assembly is listed in Table 1, and these read length distribution is shown in Fig. 1, in which there are 62.42 % reads to be above 400 bp. Using the Newbler assembly program, we generated a total of 170,248 unique sequence, comprising 28,297 contigs and 141,951 singletons with average length of 1,188 and 484 bp, respectively. The size of the assembled transcriptome is 102.4 Mbp. The N50 length of the contigs, singletons, and unique sequences was 1626, 573, and 638 bp, respectively, confirming a good quality of the assembly (Table 1). About 80.25 and 47.32 % of these contigs were >400 and >1000 bp in length (Fig. 2a). And most of these singletons (88.84 %) fell between 200 and 1400 bp in length (Fig. 2b).

Length distribution of the raw read sequence for 454 sequencing of yellow catfish

Sequence length distributions of all assembled contigs (a) and singletons (b) from yellow catfish transcriptome
Compared with recent Roche 454 pyrosequencing results, the average length of our assembled contigs (1188 bp) was much longer than that of other non-model aquaculture fish, such as Adriatic sturgeon (518 bp), turbot Scophthalmus maximus (626 bp), Bream Megalobrama pellegrini (847 bp), rainbow trout O. mykiss (758 bp), and blunt snout bream Megalobrama amblycephala (758 bp) (Salem et al. 2010; Ribas et al. 2013; Vidotto et al. 2013; Wang et al. 2012; Gao et al. 2012).
Gene Annotation and Identification of Signal Pathways Functionally Related to Reproduction, Sex Determination, and Differentiation
BLASTX searches against the NCBI non-redundant protein database (nr) led a total of 52,564 unique sequences including 18,748 contigs and 33,816 singletons, in which they were further matched to 25,669 known or predicted unique proteins (Table S1) after multiple contigs and singletons matched to the same protein were removed by practical extraction and report language. EMBOSS software analysis observed 18,295 contigs with longer than 400 bp in the putative open reading frames (ORFs) (Figure S1). To determine sequence completeness, we further calculated the ortholog-hit ratio by comparing the assembled sequence with its top hit protein, in which 2.9 % (539) of all annotated contigs and 8.3 % (2817) of all annotated singletons were revealed to have putative full-length transcripts based on the ortholog hit ratio of 1 as a full-length transcript (Zeng et al. 2013).
Gene ontology (GO) was used to describe the function of above annotated unique sequences based on three categories: biological process, cellular component, and molecular function (Fig. 3). The GO analysis assigned 11,464 (21.81 %) of the annotated unique sequences to at least one GO term. In the cellular component category, a majority of annotated unique sequences were assigned to cell (6604 sequences, GO:0005623) and cell parts terms (6604 sequences, GO:0044464). Binding (6795 sequences, GO:0005488) and catalytic activity (4914 sequences, GO:0003824) were the most represented terms within the molecular function category. In the biological process category, cellular process (7741 sequences, GO:0009987) and metabolic process (6254 sequences, GO:0008152) were the most represented terms. Reproductive system was represented by terms of reproduction (378 sequences, GO:0000003) and reproductive process (334 sequences, GO:0022414) (Table S2), from which we found many well-known sex determination and differentiation-related genes in fish, such as Dazl (Deleted in azoospermia-like) (Peng et al. 2009), Sf1 (STEROIDOGENIC factor 1) (Vizziano-Cantonnet et al. 2011), dnd (dead end) (Slanchev et al. 2005), ER-beta (estrogen receptor beta) (Vizziano-Cantonnet et al. 2011), spindlin (Sun et al. 2010), and piwi-like (P-element induced wimpy testis like) (Li et al. 2012). In 1-year-old yellow catfish (males mature about 2 years old), we observed that YY super-male showed more mature sperm cells at the periphery of tubule than XY male (data not shown). Male reproductive genes, particularly those regulating spermatogenesis, are of particular interest for us to study sex differentiation mechanisms.

Functional classification of assembled unique sequences based on gene ontology (GO) terms: molecular function, cellular component, and biological process
Subsequently, we used COG (Clusters of Orthologous Groups of proteins) for functional classification of the unique sequences. Six thousand twenty-three unique sequences were found to be involved in 24 COG categories (Fig. 4). The most enriched categories included general function prediction (2061, 24.04 %), signal transduction mechanisms (844, 9.84 %), transcription (815, 9.5 %), replication, recombination, and repair (799, 9.32 %), and posttranslational modification, protein turnover, and chaperones (642, 7.49 %). However, we did not find any sequences associated with “extracellular structures”. Moreover, we mapped 23,678 unique sequences to 320 reference canonical pathways by the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Table S2). Metabolic pathway was the largest group (2679 unique sequences, 11.31 %). Additionally, 324, 58, 41, 122, and 330 unique sequences were assigned to GnRH (gonadotropin-releasing hormone) signaling pathway, steroid hormone biosynthesis, steroid biosynthesis, ovarian steroidogenesis and estrogen signaling pathway, respectively. As shown in Fig. 5, most kinases involved in the GnRH pathway were discovered in our transcriptome, such as EGFR, CaMK, Src, Ras, and Erk1/2.

Functional classification of assembled unigenes based on Cluster of Orthologous Groups (COG) tools

Putative GnRH pathway constructed by KEGG pathway analysis. Genes identified from the transcriptome of yellow catfish are shown in red boxes. Blue boxes represent the KEGG Orthology (KO) entries
The reproductive system is generally controlled by the hypothalamus–pituitary–gonad (HPG) axis (Vadakkadath Meethal and Atwood 2005), and hypothalamic GnRH regulates synthesis and secretion of pituitary gonadotropins—LH (luteinizing hormone) and FSH (follicle stimulating hormone), and thereby stimulate synthesis of steroid sex hormones (estrogens and androgens) in the gonad, and finally induce spermatogenesis and oogenesis (Jeong and Kaiser 2006; Li et al. 2005). Embryonic GnRH signaling has been shown to be essential for maturation of the male reproductive axis (Wen et al. 2010). Steroid and steroid hormones have been extensively studied during sex determination and differentiation in vertebrates (Ramsey and Crews 2009; Nakamura 2010, 2013). In teleosts, a close connection between sexual dimorphism, reproduction, and growth has been observed (Li and Lin 2010; Chen et al. 2013a). In transgenic common carp, inhibiting GnRH synthesis could significantly reduce gonadal development (Hu et al. 2007). Generally, generation of sterile fishes can improve fish production because the energy consumption distributed to reproduction is able to transform into body composition and body weight (Chen et al. 2013a).
Characterization and Expression of Sex Determination and Differentiation-Related Genes
Since the males grow much faster than the females in yellow catfish, it is very meaningful to identify sex determination and differentiation-related genes. Searching through our Blast, GO, and KEGG results (Table S1-S3), a total of 21 known genes to be involved in sex determination or differentiation in vertebrates (Table 2) were found for the first time in yellow catfish to have significant matches. Furthermore, we used real-time PCR to detect and compare their expression patterns in 1-year-old immature XX female ovary, XY male testis, and YY testis. As shown in Fig. 6, Sox9a2, Sf1, Vasa, and Nanos show higher expression level in XX ovary than XY and YY testis, whereas Dmrt1, Sox9a1, Piwi, and ARA-α have much higher expression in XY male testis and YY testis than that in XX ovary, and Dmrt1, Sox9a1, and Piwi seem to have higher expression in YY testis than in XY testis. The detection data confirm that these candidate genes are related to sex determination or differentiation.

Relative expression profiles of eight sex determination and differentiation-related genes in XX ovary, XY, and YY testis by qRT-PCR
Actually, some of these candidate genes, such as Dmrt1, Sox9, ARA-α, Amh, and Amhr2, had been demonstrated to be involved in fish male sexual development (Xia et al. 2007; Ijiri et al. 2008; Kamiya et al. 2012; Hattori et al. 2012; Shi et al. 2012). In medaka, Dmrt1 mutation was reserved to result in a male-to-female sex reversal after the Dmy-initiated male differentiation pathways (Masuyama et al. 2012). In patagonian pejerrey, Amhy was detected in XY embryos as early as 5 days after hatching (dah), and gonadal sex differentiation began at 4 weeks after hatching (wah) in females and 6 wah in males (Hattori et al. 2012). In yellow catfish, gonadal sex differentiation was observed at 13 dah in females and 55 dah in males (Yin et al. 2008), so that the time point of sex determination should be earlier than 13 dah. Therefore, more detailed expression analysis of the candidate genes should be performed in earlier stage yellow catfish.
Our current study additionally observed significant higher expression of Dmrt1, Sox9a1, and Piwi in YY testis than in XY testis, and the expression difference might be associated with the spermatogenesis, because YY super-males were found to have more spermatids and less spermatocytes than that in XY males (data not shown). Our laboratory have identified three pairs of allelic sex chromosome-linked markers, and these markers can be used to distinguish YY super-males, XY males, and XX females from yellow catfish (Wang et al. 2009; Dan et al. 2013). Therefore, more detailed comparative and functional assays for these sex determination-related genes will be able to be performed in XX, XY, and YY samples of yellow catfish.
SSRs, SNPs, and INDELs Identification
SSRs, SNPs, and INDELs are useful molecular markers for genetic and breeding studies. However, only a few SSR markers are available for yellow catfish until now (Wang et al. 2009; Liang et al. 2012). Through using MISA software to analyze the transcriptome, we identified 82,794 SSRs (1–6 bp repeat motif), widely distributing in 51,062 sequences, and accounting for 29.99 % of the total unique sequences (Table 3). As shown in Table 3, there are 17,878 sequences with more than 1 SSR, among which 22,218 SSRs are present in compound formation. Di-nucleotide repeats are the most frequent form of SSRs (53,933, 65.14 %), and then follow by mononucleotide repeats (14,168, 17.11 %), trinucleotide repeats (8,104, 9.79 %), tetranucleotide repeats (6,027, 7.28 %), pentanucleotide repeats (478, 0.58 %), and hexanucleotide repeats (84, 0.1 %).
Bowtie2 was used to generate aligned file, which was considered as input for SNPs and INDELs discovery using samtools, under stringent filtering criteria (covered by at least 10 reads and with a minor allelic frequency ≥20 %). A total of 26,450 SNPs including 12,755 transitions and 13,695 transversions and 4145 INDELs were identified from the unique sequences (Fig. 7). The A/G (3486), G/A (2945), C/T (3403), and T/C (2921) SNP transitions were about equally distributed. For SNPs transversion, A/T (2161) and T/A (2039) were the most common, whereas G/C (1155) and C/G (1362) were the smallest types. These differences might be largely due to the base structure and hydrogen bond interaction between these base pairs (Ma et al. 2012).

Classification and distribution of putative SNPs and INDELs identified from 454 transcriptome of yellow catfish
SSRs, SNPs, and INDELs have been widely applied into genetic breeding studies in aquaculture animals. Our current identified SSRs and SNPs will be able to apply to diverse studies on parentage detection, population evaluation, quantitative trait locus (QTL) mapping, and phylogenetic analysis in yellow catfish. If more samples are assayed, we will be able to screen higher polymorphic SNPs and to construct a SNP-based high-density genetic map.
Conclusion
In this study, we performed de novo transcriptome sequencing to provide an accurate assembly and effective gene coverage of yellow catfish. Based on the comparative transcriptome analysis of differentially expressed genes between XX ovary, XY testis, and YY testis, we acquired a more broad coverage of genes related to sex determination and differentiation, and screened at least 21 sex determination and differentiation-related genes. Moreover, their expression patterns were partially characterized in XX female ovary, XY male testis, and YY testis of yellow catfish. Additionally, a total of 82,794 SSRs, 26,450 SNPs, and 4,145 INDELs were identified from the transcriptome data.
References
Baroiller JF, D’Cotta H, Bezault E, Wessels S, Hoerstgen-Schwark G (2009) Tilapia sex determination: where temperature and genetics meet. Comp Biochem Physiol A Mol Integr Physiol 153:30–38
Chen J, Hu W, Zhu ZY (2013a) Progress in studies of fish reproductive development regulation. Chin Sci Bull 58:7–16
Chen Z, Gui J, Gao X, Pei C, Hong Y, Zhang Q (2013b) Genome architecture changes and major gene variations of Andrias davidianus ranavirus (ADRV). Vet Res 44:101
Dan C, Mei J, Wang D, Gui J (2013) Genetic differentiation and efficient sex-specific marker development of a pair of Y- and X-linked marker in yellow catfish. Int J Biol Sci 9:1043–1049
Gao Z, Luo W, Liu H, Zeng C, Liu X et al (2012) Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama amblycephala). PLoS ONE 7:e42637
Gui J (2007) Genetic basis and artificial control of sexuality and reproduction in fish. Science Press, Beijing
Gui J, Zhou L (2010) Genetic basis and breeding application of clonal diversity and dual reproduction modes in polyploid Carassius auratus gibelio. Sci China Life Sci 53:409–415
Gui J, Zhu Z (2012) Molecular basis and genetic improvement of economically important traits in aquaculture animals. Chin Sci Bull 57:1751–1760
Haffray P, Vauchez C, Vandeputte M, Linhart O (1998) Different growth and processing traits in males and females of European catfish, Silurus glanis. Aquat Living Resour 11:341–345
Hattori RS, Murai Y, Oura M, Masuda S, Majhi SK et al (2012) A Y-linked anti-Mullerian hormone duplication takes over a critical role in sex determination. Proc Natl Acad Sci U S A 109:2955–2959
Henken A, Brunink A, Richter C (1987) Differences in growth rate and feed utilization between diploid and triploid African catfish, Clarias gariepinus (Burchell 1822). Aquaculture 63:233–242
Hu W, Li SF, Tang B, Wang YP, Lin HR et al (2007) Antisense for gonadotropin-releasing hormone reduces gonadotropin synthesis and gonadal development in transgenic common carp (Cyprinus carpio). Aquaculture 271:498–506
Huang F, Yan A, Xiong C, Zhen R, Zhang G (1999) Evaluation of the nutrition and the rate of flesh in the whole body of Pelteobagrus fulvidraco rich. Freshw Fish 29:3–6
Huang W, Zhou L, Li Z, Gui JF (2009) Expression pattern, cellular localization and promoter activity analysis of ovarian aromatase (Cyp19a1a) in protogynous hermaphrodite red-spotted grouper. Mol Cell Endocrinol 307:224–236
Ijiri S, Kaneko H, Kobayashi T, Wang DS, Sakai F et al (2008) Sexual dimorphic expression of genes in gonads during early differentiation of a teleost fish, the Nile tilapia Oreochromis niloticus. Biol Reprod 78:333–341
Jeong K, Kaiser U (2006) Gonadotropin-releasing hormone regulation of gonadotropin biosynthesis and secretion. In: Neill JD (ed) Knobil and Neill’s physiology of reproduction. Elsevier, Amsterdam, pp 1635–1726
Kamiya T, Kai W, Tasumi S, Oka A, Matsunaga T et al (2012) A trans-species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu). PLoS Genet 8:e1002798
Kobayashi Y, Nagahama Y, Nakamura M (2013) Diversity and plasticity of sex determination and differentiation in fishes. Sex Dev 7:115–125
Lee BY, Coutanceau JP, Ozouf-Costaz C, D’Cotta H, Baroiller JF et al (2011) Genetic and physical mapping of sex-linked AFLP markers in Nile tilapia (Oreochromis niloticus). Mar Biotechnol (NY) 13:557–562
Li CJ, Zhou L, Wang Y, Hong YH, Gui JF (2005) Molecular and expression characterization of three gonadotropin subunits common alpha, FSHbeta and LHbeta in groupers. Mol Cell Endocrinol 233:33–46
Li M, Hong N, Gui J, Hong Y (2012) Medaka piwi is essential for primordial germ cell migration. Curr Mol Med 12:1040–1049
Li WS, Lin HR (2010) The endocrine regulation network of growth hormone synthesis and secretion in fish: Emphasis on the signal integration in somatotropes. Sci China Life Sci 53:462–470
Li XY, Zhang XJ, Li Z, Hong W, Liu W, Zhang J, Gui JF (2014a) Evolutionary history of two divergent Dmrt1 genes reveals two rounds of polyploidy origins in gibel carp. Mol Phylogenet Evol 78:96–104
Li XY, Li Z, Zhang XJ, Zhou L, Gui JF (2014b) Expression characterization of testicular DMRT1 in both Sertoli cells and spermatogenic cells of polyploid gibel carp. Gene 548:119–125
Liang HW, Hu GF, Li Z, Zou GW, Liu XL (2012) Mitochondrial DNA sequence of yellow catfish (Pelteobagrus fulvidraco). Mitochondrial DNA 23:170–172
Liao X, Cheng L, Xu P, Lu G, Wachholtz M et al (2013) Transcriptome analysis of Crucian Carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS ONE 8:e62308
Liu H, Cui S, Hou C, Xu J, Chen H (2007) YY super-male generated gynogenetically from XY female in Pelteobagrus fulvidraco (Richardson). Acta Hydrobiol Sin 31:718–725
Liu F, Sun F, Li J, Xia JH, Lin G et al (2013a) A microsatellite-based linkage map of salt tolerant tilapia (Oreochromis mossambicus x Oreochromis spp.) and mapping of sex-determining loci. BMC Genomics 14:58
Liu H, Guan B, Xu J, Hou C, Tian H et al (2013b) Genetic manipulation of sex ratio for the large-scale breeding of YY super-male and XY all-male yellow catfish (Pelteobagrus fulvidraco (Richardson)). Mar Biotechnol (NY) 15:321–328
Ma K, Qiu G, Feng J, Li J (2012) Transcriptome analysis of the oriental river prawn, Macrobrachium nipponense using 454 pyrosequencing for discovery of genes and markers. PLoS ONE 7:e39727
Masuyama H, Yamada M, Kamei Y, Fujiwara-Ishikawa T, Todo T et al (2012) Dmrt1 mutation causes a male-to-female sex reversal after the sex determination by Dmy in the medaka. Chromosome Res 20:163–176
Matsuda M, Nagahama Y, Shinomiya A, Sato T, Matsuda C et al (2002) DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature 417:559–563
Mei J, Gui JF (2014) Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Sci China Life Sci 57: in press
Mei J, Yue HM, Li Z, Chen B, Zhong JX et al (2013) C1q-like Factor, a Target of miR-430, Regulates Primordial Germ Cell Development in Early Embryos of Carassius auratus. Int J Biol Sci 10:15–24
Nakamura M (2010) The mechanism of sex determination in vertebrates-are sex steroids the key-factor? J Exp Zool A Ecol Genet Physiol 313:381–398
Nakamura M (2013) Is a sex-determining gene(s) necessary for sex-determination in amphibians? Steroid hormones may be the key factor. Sex Dev 7:104–114
Peng JX, Xie JL, Zhou L, Hong YH, Gui JF (2009) Evolutionary conservation of Dazl genomic organization and its continuous and dynamic distribution throughout germline development in gynogenetic gibel carp. J Exp Zool B Mol Dev Evol 312:855–871
Ramsey M, Crews D (2009) Steroid signaling and temperature-dependent sex determination-Reviewing the evidence for early action of estrogen during ovarian determination in turtles. Semin Cell Dev Biol 20:283–292
Ribas L, Pardo BG, Fernandez C, Alvarez-Dios JA, Gomez-Tato A et al (2013) A combined strategy involving Sanger and 454 pyrosequencing increases genomic resources to aid in the management of reproduction, disease control and genetic selection in the turbot (Scophthalmus maximus). BMC Genomics 14:180
Salem M, Rexroad CE 3rd, Wang J, Thorgaard GH, Yao J (2010) Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 11:564
Sarder MR, Penman DJ, Myers JM, McAndrew BJ (1999) Production and propagation of fully inbred clonal lines in the Nile tilapia (Oreochromis niloticus L.). J Exp Zool 284:675–685
Shi Y, Liu X, Zhang H, Zhang Y, Lu D et al (2012) Molecular identification of an androgen receptor and its changes in mRNA levels during 17alpha-methyltestosterone-induced sex reversal in the orange-spotted grouper Epinephelus coioides. Comp Biochem Physiol B Biochem Mol Biol 163:43–50
Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL et al (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346:240–244
Slanchev K, Stebler J, de la Cueva-Mendez G, Raz E (2005) Development without germ cells: the role of the germ line in zebrafish sex differentiation. Proc Natl Acad Sci U S A 102:4074–4079
Smith CA, Roeszler KN, Ohnesorg T, Cummins DM, Farlie PG et al (2009) The avian Z-linked gene DMRT1 is required for male sex determination in the chicken. Nature 461:267–271
Sun M, Li Z, Gui JF (2010) Dynamic distribution of spindlin in nucleoli, nucleoplasm and spindle from primary oocytes to mature eggs and its critical function for oocyte-to-embryo transition in gibel carp. J Exp Zool A Ecol Genet Physiol 313:461–473
Taranger GL, Carrillo M, Schulz RW, Fontaine P, Zanuy S et al (2010) Control of puberty in farmed fish. Gen Comp Endocrinol 165:483–515
Vadakkadath Meethal S, Atwood CS (2005) The role of hypothalamic-pituitary-gonadal hormones in the normal structure and functioning of the brain. Cell Mol Life Sci 62:257–270
Vidotto M, Grapputo A, Boscari E, Barbisan F, Coppe A et al (2013) Transcriptome sequencing and de novo annotation of the critically endangered Adriatic sturgeon. BMC Genomics 14:407
Vizziano-Cantonnet D, Anglade I, Pellegrini E, Gueguen MM, Fostier A et al (2011) Sexual dimorphism in the brain aromatase expression and activity, and in the central expression of other steroidogenic enzymes during the period of sex differentiation in monosex rainbow trout populations. Gen Comp Endocrinol 170:346–355
Wang D, Mao HL, Chen HX, Liu HQ, Gui JF (2009) Isolation of Y- and X-linked SCAR markers in yellow catfish and application in the production of all-male populations. Anim Genet 40:978–981
Wang J, Yu X, Zhao K, Zhang Y, Tong J et al (2012) Microsatellite development for an endangered bream Megalobrama pellegrini (Teleostei, Cyprinidae) using 454 sequencing. Int J Mol Sci 13:3009–3021
Wen S, Ai W, Alim Z, Boehm U (2010) Embryonic gonadotropin-releasing hormone signaling is necessary for maturation of the male reproductive axis. Proc Natl Acad Sci U S A 107:16372–16377
Xia W, Zhou L, Yao B, Li CJ, Gui JF (2007) Differential and spermatogenic cell-specific expression of DMRT1 during sex reversal in protogynous hermaphroditic groupers. Mol Cell Endocrinol 263:156–172
Xiao Q, Xia JH, Zhang XJ, Li Z, Wang Y, Zhou L, Gui JF (2014) Type-IV antifreeze proteins are essential for epiboly and convergence in gastrulation of zebrafish embryos. Int J Biol Sci 10:715–732
Xu S, Xia W, Zohar Y, Gui JF (2013) Zebrafish dmrta2 regulates the expression of cdkn2c in spermatogenesis in the adult testis. Biol Reprod 88:14
Yano A, Guyomard R, Nicol B, Jouanno E, Quillet E et al (2012) An immune-related gene evolved into the master sex-determining gene in rainbow trout, Oncorhynchus mykiss. Curr Biol 22:1423–1428
Yin HB, Jia ZH, Yao DX, Sun ZW, Yu B et al (2008) Sex differentiation in Pelteobagrus fulvidraco. Chin J Zool 43:103–108
Zeng V, Ewen-Campen B, Horch HW, Roth S, Mito T et al (2013) Developmental gene discovery in a hemimetabolous insect: de novo assembly and annotation of a transcriptome for the cricket Gryllus bimaculatus. PLoS ONE 8(5):e61479
Zhou L, Gui JF (2010) Molecular mechanisms underlying sex change in hermaphroditic groupers. Fish Physiol Biochem 36:181–193
Acknowledgments
This work was supported by grants to Jie Mei from the Fundamental Research Funds for the Central Universities (2013PY068, 52902–0900202496, 52204–12018) and the National Natural Science Foundation of China (31301931), and to Jian-Fang Gui from the special Fund for Agro-scientific Research in the Public Interest from the Ministry of Agriculture of China (2009030406), the National Key Basic Research Program (2010CB126301). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
Length distribution of Open reading frame (ORF) from all contigs of the transcriptome. (GIF 7 kb)
Table S1
Summary of BLAST nr result for all unique sequences in the transcriptome. (XLSX 1404 kb)
Table S2
List of all the reproduction and reproductive process related genes found in GO analysis of the yellow catfish transcriptome. (XLSX 107 kb)
Table S3
KEGG pathways discovered in the unigenes, contigs and singletons of yellow catfish transcriptome (XLSX 790 kb)
Table S4
The primer sequences for real-time PCR study. (DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Chen, X., Mei, J., Wu, J. et al. A Comprehensive Transcriptome Provides Candidate Genes for Sex Determination/Differentiation and SSR/SNP Markers in Yellow Catfish. Mar Biotechnol 17, 190–198 (2015). https://doi.org/10.1007/s10126-014-9607-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-014-9607-7