
Abstract
Brucella abortus and Brucella melitensis are the primary etiological agents of brucellosis in large and small ruminants, respectively. There are limited comparative genomic studies involving Brucella strains that explore the relatedness among both species. In this study, we involved strains (n=44) representing standard, vaccine and Indian field origin for pangenome, single nucleotide polymorphism (SNP) and phylogenetic analysis. Both species shared a common gene pool representing 2884 genes out of a total 3244 genes. SNP-based phylogenetic analysis indicated higher SNP diversity among B. melitensis (3824) strains in comparison to B. abortus (540) strains, and a clear demarcation was identified between standard/vaccine and field strains. The analysis for virulence genes revealed that virB3, virB7, ricA, virB5, ipx5, wbkC, wbkB, and acpXL genes were highly conserved in most of the Brucella strains. Interestingly, virB10 gene was found to have high variability among the B. abortus strains. The cgMLST analysis revealed distinct sequence types for the standard/vaccine and field strains. B. abortus strains from north-eastern India fall within similar sequence type differing from other strains. In conclusion, the analysis revealed a highly shared core genome among two Brucella species. SNP analysis revealed B. melitensis strains exhibit high diversity as compared to B. abortus strains. Strains with absence or high polymorphism of virulence genes can be exploited for the development of novel vaccine candidates effective against both B. abortus and B. melitensis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bovine brucellosis is a contagious and serious disease of livestock associated with significant economic losses due to reduced livestock production and reproductive performances. The zoonotic nature of pathogen also poses a potential threat to public health. The disease is termed as Bangs disease or contagious abortion and undulant or intermittent fever in animals and humans, respectively (Megid et al. 2010). Brucellosis is caused by the Gram-negative bacteria Brucella, which belongs to the family Alphaproteobacteria. The genus Brucella includes multiple species, traditionally grouped as classical and non-classical species. Six classical species predominantly associated with mammals are B. melitensis, B. abortus, B.canis, B. suis, B. ovis, and B. papionis. Among the non-classical species, B. microti and B. pinnipedalis are primarily known for infections in land and marine mammals (Guzman-Verri et al. 2020). Among all, B. abortus, B. melitensis, and B. suis are the pathogens of zoonotic and public health importance, out of which B. melitensis cause serious infections in humans as compared to others. On the other hand, B. abortus secreted in milk, milk products and body fluids can be contracted to the humans via consumption or close contact (Halling et al. 2005).
Vaccines are commercially available for the control of brucellosis in ruminants. The vaccines against brucellosis generally employs live attenuated strains capable of inducting Th1 immune response, protection against uterine colonization, and systemic infection and exhibits no interference with the serodiagnosis (Schuriget al. 1995). Bovine brucellosis can be controlled by vaccination using B. abortus S19 and B. abortus RB51 vaccine strains. In small ruminant, B. melitensis Rev.1 strain is used as the vaccine candidate. All the three vaccine strains are recognized as naturally attenuated strains (Yang et al. 2013). Continuous monitoring of the herds based on clinical signs and serodiagnosis is indispensable for the control of brucellosis. Serodiagnosis in developing countries is routinely performed using assays such as Rose Bengal plate test and standard tube agglutination tests. However, these assays cannot differentiate the species of Brucella involved. Molecular methods, primarily polymerase chain reaction and gene sequencing methods, have gained wider acceptance and can achieve differentiation up to the biovar levels. Recently, whole-genome sequencing–based approaches (single nucleotide polymorphism (SNP)–based and core genome multilocus sequence typing (MLST)) are proposed for better strain typing (Uelze et al. 2020; Abdel-Glil et al. 2022). WGS analysis of isolates from different geographical regions may give insight into the diversity of strains involved in infections (Karthik et al. 2021).
India is one among the several Asian countries where brucellosis is considered endemic. However, limited studies have been carried out for the genomic characterization of Brucella isolates. Irrespective of the genomic similarity within B. abortus and B. melitensis, they have the capability to evolve differently, envisaging robust approaches for strain characterization (Guzman-Verri et al. 2020). Importantly, there is a critical requirement to set up a genome sequence–based standards for vaccine, outbreak, and reference strains (Wang et al. 2019). So a comparative genomics study involving standard, vaccine and field strains will offer the possibility of exploring gene compositions, SNP, and genotypes among related species and strains (Hurtado et al. 2020; Bengtsson et al. 2022). Determining species-specific and shared pangenomes can suggest genetics of the established vaccine strains and its derivatives (Bogaards et al. 2019). With this background, the present study envisaged a comparative genomic analysis of field strains reported in India along with standard/vaccine strains of B. abortus and B. melitensis.
Materials and methods
Genome data retrieval and genome feature analysis
High-quality genome sequences of Indian field strains of B. abortus (n=22) and B. melitensis (n=15), standard (reference) strains (BA_544 (GCA_000369945.1), BA_RB51 (GCA_000366025.1), BA_2308(GCA_000054005.1) and BM_16M (GCF_000740415.1)) and vaccine strains (BA_S19 (GCA_000018725.1), BA_S19 deltaper (GCA_001038665.1) and BM_Rev1 (GCA_002953595.1)) were downloaded from the BV-BRC database using batchentrez (https://www.ncbi.nlm.nih.gov/sites/batchentrez) following quality filtrations (https://www.bv-brc.org). The metadata of the downloaded genomic assemblies are provided in the Table S1. The assemblies were analyzed for virulence and AMR genes, prophage elements, and genomic islands. For virulence and AMR gene predictions, the genome assemblies were subjected to ABRicate (v.1.0.1) searches against the virulence factor database (VFDB) (Liu et al. 2019) and CARD 3.0.3 database (Alcock et al. 2020). The prediction parameters were set for a minimum DNA identity and coverage of 80% and were carried out within the galaxy web server (https://usegalaxy.eu). Prophage elements in the genomes were predicted using PHASTER (https://phaster.ca/) and genomic islands were predicted using Island Viewer 4 (Bertelli et al. 2017). Prophage and genomic island predictions were carried out for the complete genome sequence of B. abortus (BA_2308) and B. melitensis (BM_16M) strains. Further, the predicted prophages and genomic islands were mapped to BLAST atlas employing all the genome sequences using BRIG tool to unravel their sharing or unique patterns (Alikhan et al. 2011).
Pangenome and SNP analysis
Pangenome analysis was carried out following Prokka v 1.14.6 annotation (Seemann 2014) using Panaroo v.1.3.0 (Tonkin-Hill et al. 2020). Panaroo parameters were set to the strict mode that enabled the removal of invalid genes. Two-dimensional scaling and visualization of pangenome was performed using FriPan (https://github.com/drpowell/FriPan). From the pool of total genes, the shared and unique genes of both species (B. abortus and B. melitensis) were inferred. The species-specific unique genes were scanned for COGs annotations in eggnog-mapper tool (http://eggnog-mapper.embl.de).
The SNPs in the genomes were identified using the KSNP3 package (Gardner et al. 2015). Within the package, the optimal kmer and FCK (the fraction of core kmers) sizes were calculated using the Kchooser script. For annotating SNPs, the strains BA_2308 and BM_16M were used as reference. The KSNP3 tool was used to create the SNP matrix, and SNP-based tree (tree_AlleleCounts.ML.NodeLabel.tre). The internal nodes were labeled with the node number followed by the number of SNPs that are shared exclusively by the descendants of that node separated by an underscore. The tree figure was visualized using Dendroscope V3.8.4 (Huson et al. 2012).
Whole-genome phylogeny and multilocus sequence typing analysis
Phylogenetic analysis was carried out using the whole-genome sequences of B. abortus and B. melitensis strains. A multiple genome alignment was generated using the realphy webserver (https://realphy.unibas.ch/realphy/).The phylogenetic tree was constructed based on the alignment file (*.FASTA) using the IQTREE software (Trifinopoulos et al. 2016) applying the GTR+G substitution model and maximum likelihood methodology (1000 bootstraps). The generated phylogenetic tree was visualized and annotated using the iTOL v5 web tool (Leutnic and Bork 2021).
Retrieved genome assemblies were subjected to MLST analysis using the PubMLST database. Identification of sequence types (STs) was carried out using the 9, 21, and cgMLST schemes as described earlier (Jolley et al. 2018; Abdel-Glil et al. 2022). Brucella spp. (cgMLST) scheme was downloaded from the cgMLST.org, Nomenclature Server (https://www.cgmlst.org/ncs), and the analysis was carried out using the PyMLST V2.1 tool (https://github.com/bvalot/pyMLST). For this, a database was created followed by the addition of genomic data of all the strains. Finally, a matrix of cgMLST distance was defined as the number of different alleles between each pair of two strains, omitting the missing data. The matrix file was used to create a minimum spanning tree (MST) using MSTree V2 option in GrapeTree (Zhou et al. 2018).
Results
Genome features, AMR, prophages and genomic islands
The genomic size of the strains included in the study was ranging from 3.2 to 3.3 Mb with a conserved GC content of 57.2%, whereas the CDS number ranged from 3350 to 3800. The genomic features of all assemblies including the size, GC content, number of contigs, tRNA, rRNA, and CDS are provided in Table 1. No AMR genes were predicted among the strains, except for the genes such as aph(3′)-Ia and aac(3′)-Ia coding for aminoglycoside phosphotransferase and acetyl transferase, respectively, in the BA_S19 deltaper and BA_ML7, strains respectively.
A total of 26 and 23 genomic islands were predicted in the B. abortus and B. melitensis genomes, respectively. Details of genomic islands predicted for both species are provided in the Table S2. An incomplete prophage of 17.4Kb was predicted in the B. melitensis BM_16M strain, whereas no prophage was predicted in the B. abortus strain BA_2308. BRIG analysis indicated high conservation of genomic islands and prophages for both the species. Other than this, a unique genomic region was identified in the BM_16M strain and was shared with some of B. melitensis strains (BM_LM17, BM_LM18, BM_LM19, and BM_LM20), but not with any of the B. abortus strains (Fig. 1). This unique region (approx. 20 kb) was identified to be carrying various genes associated with sugar metabolism.

Comparative alignment of Brucella genomes. BRIG analysis indicated highly conserved genomic islands and prophages for both the species
Pangenome analysis of B. abortus and B. melitensis
Pangenome analysis of the 44 strains involving B. abortus (n =27) and B. melitensis (n=17) strains identified 3244 genes representing total number of orthologous genes, of which 2884 were core (shared by all strains from both species) and the remaining 360 were accessory (shared by few strains or unique to any strain) genes, respectively. This indicates a higher-level conservation of genes among both the Brucella species. Multidimensional scaling revealed two major groups, one each for B. abortus and B. melitensis, and these two groups differed by approximately 120 genes. B. abortus strains LMN1 and LMN2 were distantly clustered, indicative of unique accessory gene composition (Fig. 2).

Pangenomic analysis of Brucella strains A Multidimensional scaling plot B Gene presence/absence tree C Pan-genome overview
Functional annotation was carried out for the accessory genes. A total of 149 CDS were identified as accessory genes in B. abortus, of which functional predictions were only available for 53 CDS genes. Prophage elements were predicted for majority of the accessory CDS. Among these fifty-three CDS, most were encoding cell wall hydrolase (N-acetylmuramyl-l-alanine amidase), acyl transferase, and methyl transferase enzymes. In addition, plasmid partition protein and nucleosome segregation protein along with phage-associated proteins were also identified. The summary of accessory genes predicted in the B. abortus genomes are provided in Table S3. Accessory genes in the B. melitensis were associated with carbohydrate metabolism (polysaccharide synthesis, transfer, and modification). DNA invertase and site-specific recombinase were also predicted in the B. melitensis isolates. The genomes also contain region coding for proteins related to glycosylphosphatidylinositol synthesis and outer membrane proteins. Small transmembrane protein TraB orthologs were also identified as part of accessory genes in the B. melitensis isolates. The summary of accessory genes predicted in the B. melitensis genomes is provided in Table S4.
Single nucleotide polymorphism analysis
The KSNP3 analysis found 8921 SNPs, of which 2623 were synonymous and 4734 were non-synonymous. Additional 1565 SNPs were not representing any of the CDS regions identified in the annotated genomes. Following genome comparison, the NS/S ratio was found to be 1.8, indicating that the population is evolving and becoming more diversified. In the SNP-based phylogeny (Figure S1), B. melitensis and B. abortus strains were divided into two clusters. B. melitensis isolates exclusively shared 3824 SNPs (node 20), but isolates of B. abortus shared considerably fewer SNPs (540) (node 14). This indicates that the B. melitensis strains are undergoing more diversification as compared to the B. abortus strains.
B. abortus strains BA_2308, BA_544, BA_RB51, BA_S19, and BA_S19 deltaper constituted distinct group with 38 exclusive SNPs (node 15). Very few SNPs were shared by other Indian B. abortus strains. However, 44 SNPs were exclusively shared by vaccine strains BA_S19 and BA_S19 deltaper (node 16) (Figure S1). B. melitensis strains BM_16M and BM_Rev 1 involving node (node 21) shared 1171 SNPs exclusively and formed an outgroup from the field B. melitensis strains. A total of 1004 SNPs were shared by the remaining Indian B. melitensis strains.
Synonymous SNPs were observed in several genes coding for proteins such as transcription-repair coupling factor, entericidin A/B family lipoprotein, DNA polymerase III subunit epsilon, and flagellar hook protein FlgE. On the other hand, nonsynonymous SNPs indicative for amino acid changes were mostly observed in membrane-associated proteins such as transporters. This included the porins, autotransporter outer membrane beta-barrel, PAS domain S-box protein, bacteriophage abortive infection AbiH family, carbohydrate ABC transporter permease, heavy metal translocating P-type ATPase, excinuclease ABC subunit UvrB, and Tm-1-like ATP-binding domain-containing protein, respectively. Details of SNPs (non-synonymous and synonymous) identified in the comparative SNP analysis of the44 strains are provided in Table S5.
Virulence factors
The presence of genes coding for 43 virulence factors was searched within the Brucella genomes. Majority of the isolates were found to be carrying all the analyzed genes. Exceptions were observed with the absence of certain virulence factors in the strains such as BM_2007 BM1 (wbkA and ricA), BA_S19 deltaper (wbkB), and BA_RB51 (wboA). Among these, S19 deltaper is a targeted mutant strain for the wbkB gene. Along with that, most of the virulence factors showed 100% consensus with the reference sequences in VFDB database. The analysis revealed that the virulence genes virB3, virB7, ricA, virB5, ipx5, wbkC, wbkB, and acpXL were highly conserved among all the strains. The genes fabZ, gmd, lpxC, per, virB, wzm, and wbkA were largely conserved among the B. melitensis isolates. All B. abortus strains, along with the B. melitensis strains BM_16M and BM_rev.1, shared a conserved ManAoAg gene, whereas this gene was found to be exhibiting polymorphisms in other B. melitensis strains. Forty-one strains involved in the study had virB2 gene with moderate sequence variability; however, B. abortus strains had virB10 gene with highest variability among all the 43 virulence factor genes (>14%) (Figure S2).
Whole-genome phylogeny and MLST analysis
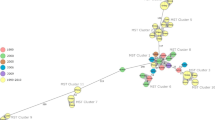
Two distinct clusters of B. abortus and B. melitensis were evident in the whole genome–based phylogeny. The standard strains (BM_16M and BM_Rev.1) formed separate subcluster separated from the other Indian strains within the B. melitensis cluster. Similar findings were observed with the B. abortus strains where the standard/vaccine strains and field strains from India formed separate subclusters. The genotypes of standard and Indian strains were evaluated using MLST analysis based on 9 locus, 21 locus, and cgMLST schemes. It was observed that the sequence types and whole genome–based phylogeny have good correlations. Standard isolates of B. melitensis belonged to ST-7 and ST-73 with 9 and 21 locus MLST, respectively. However, the field strains of B. melitensis were found to belong to the ST-8 based on both MLST schemes. The laboratory maintained B. abortus strains such as BA_2308, BA_RB51, BA_S19, and BA_S19 deltaper belonged to ST-5, whereas BA_544 strain was represented as ST-1 genotype. Indian strains were found to be the closest or precise matches for ST-1. In comparison to the other two MLST systems, cgMLST had more discriminatory features. The isolates from the north-eastern region of India formed subclusters and shared similar genotypes (closest to ST-563 or 564). This indicates that a similar type of B. abortus strains circulates in the North East region of India. According to the cgMLST scheme, the BA_S19 deltaper mutant strain ST represented a profile nearest to ST-223 or 811. The S19 deltaper mutant strain exhibits four locus variants as compared to BA_S19 strain, whereas original strain BA_S19 was designated as genotype ST-223. This indicates that high strain typing efficiency can be achieved with cgMLST scheme and can differentiate strains with limited genetic variations. The Indian strain BA_85/69 placed as an outgroup exhibited distinct MLST profile (9 locus (31), 21 locus (63), cgMLST (361)) (Fig. 3). The MST analysis of cgMLST distance matrix revealed the clustering of two species separately. Within each species, standard strains formed a distant subcluster from the field strains (Fig. 4).

Whole genome phylogenetic analysis. Comparison of whole-genome phylogenetic tree and MLST sequence types

Minimum spanning trees (MST) based on cgMLST distance matrix A MST highlighting B. abortus and B. melitensis strains B MST highlighting field, vaccine and pathogenic strains
Discussion
Bovine brucellosis is a contagious disease of livestock sector and a potent zoonosis. The study analyzed the whole-genome sequence data of B. abortus and B. melitensis strains representing standard, vaccine and field origins. Genome assembly and annotation statistics indicated limited variations in genome size, GC content and CDS number among the involved strains (Table 1). Earlier studies reported significant degree of similarity for Brucella species based on 16sRNA and phylogenetic analysis (Foster et al. 2009). In India, B. abortus S19 live attenuated strain is currently used as the vaccine candidate for the control of large ruminant brucellosis. However, the protection level provided by B. abortus S19 strain against other Brucella infections is unknown (Van straten et al. 2016). For inferring such a possibility, the preliminary step is to understand the genetic similarity and uniqueness of both species with respect to standard, vaccine and field strains. A pangenome analysis involving the B. abortus and B. melitensis strains indicated 3244 total genes, of which 2884 genes were core, representing genes that are shared by all the strains from both species (Fig. 2). A high core vs pangenome ratio of 88% (2884/3244) shows both species harbor very little unique genes and accounts for around 120 genes in each species (Table S3 and S4). Accessory genome and strain-specific unique genetic elements are usually carried in plasmids, prophages, and genomic islands in bacterial genomes (Ozer et al. 2014). Both species were indicated for genomic islands and prophages that were conserved across species (Table S2). However, a unique 20-kb region was found in few B. melitensis strains but not in B. abortus strains. Exceptional presence of unique accessory genes observed in the two B. abortus strains (LMN1 and LMN) were found to be associated with phage proteins (Fig. 2). Previous studies also identified certain B. melitensis strains of Indian origin for their unique accessory gene content (Karthik et al. 2021). However, the overall analysis indicated highly conserved genomes and significantly shared core genome among both species. This homology indicate the probability for a similar mechanisms in both species contributing towards immune response following vaccination or infections. Hence, an appropriate vaccine candidate derived from one species may evoke a significant immune response effective against Brucella infection mediated by a different species.
Further, the extent of divergence between both species attributed to SNPs was evaluated. SNPs can define genetic variations occurring in strains that can also attribute to phenotypic characteristics associated with invasion, survival, and virulence. Alignment and reference-free SNP detection tool were used for this (Gardner et al. 2015). SNP matrix indicated number of SNP difference between both species. B. melitensis were found to evolve more quickly and had a higher number of SNPs in their genomes. B. abortus strains showed less divergence based on SNP variations (Figure S1, Table S5). Earlier studies have analyzed the SNPs associated with the genomes of Brucella strains across the world, and the results showed the establishment of genotypes and sub-genotypes depending on their SNP profiles (Pisarenko et al. 2018). BM_16M and BM_Rev.1 strain showed major difference in SNP profile from other Indian strains. On the other hand, B. abortus vaccine candidate strains showed lesser number of exclusive SNPs shared as compared to B. melitensis vaccine strains. It may be possible that some of these involved SNPs are important in the attenuation of candidate vaccine strains (Kornspan et al. 2020). Thus, by using a genomic approach, Brucella field and vaccine strains can be distinguished based on their unique SNP profiles (El-Sayedet al. 2018). On the other hand, gene-based approach may not be suitable to identify closely related strains (Tan et al. 2015). Whole-genome phylogeny (WGP)-SNP-based phylogeny can discriminate the strains based on the regions. The clustering of isolates from the Northeastern region indicates that strains belonging to same genotypes may be circulating in this area. This is supported by the previous findings where Brucella strains from the different South African regions were found clustering as different genotypes with specific SNPs (Ledwaba et al. 2021). The importance of attenuation and decreased virulence can be inferred from the vaccine strains with the highest number of non-synonymous SNPs (NS) found. When the S19 genome was compared to virulent genomes, Orf and amino acid alterations were also identified in the S19 genome along with eryC gene deletion (Crasta et al. 2008). NS mutation in the YadA protein affects the adherence, invasiveness, and serum resistance capacity of bacteria. It has structural similarity with Yersinia enterocolitica YadA protein (Paulsen et al. 2002). Similarly, SNPs in the phosphomannomutase, ABC transporter permease, and GDP-mannose dehydratase genes are presumed to have a role in the conversion of smooth to rough phenotype and attenuation in B. melitensis Rev 1 vaccine strain (Kornspan et al. 2020).
The pathogenesis of Brucella spp. in the susceptible host depends on the presence of virulence factors associated with LPS synthesis, immune evasion, intracellular survival, gene regulation, and secretion system. The present study identified a highly conserved nature of many significant genes including those coding for the virulence factors among both Brucella species. However, a degree of genetic variability was observed among certain virulence genes in many of the strains analyzed. Also, some of the strains were found to be lacking the ricA, wbkA, wbkB, and wboA genes (Figure S2). The RicA protein plays a role in the Brucella intracellular trafficking and translocation process (De Barsy et al. 2011). The wbo and wbk genes are associated with the production of the LPS O chain and codes for the enzymes glycosyltransferase (wboA) and manosyltransferase (wbkA), respectively. The gene wbkB codes for an enzyme of unknown function. Brucella strains may develop a rough phenotype when these genes, particularly (wboA), are disrupted (Etemady et al. 2015). The wbkA gene interacts with wboA gene and is involved in the elongation of the O chain. Thus, both genes may compensate each other to overcome phenotypic variations (Cloeckart et al. 2000). However, in B. melitensis 16M, the wbkB gene deletion did not change LPS composition or morphology (Godfroid et al. 2000). In contrast, B. abortus S19, a wbkB mutant, exhibited transition from smooth LPS to intermediate smooth LPS (Chaudhuriet al. 2015). Thus, the phenotypic variation associated with the gene mutation may vary with species or strains. Certain genetic elements such as ManAoAg gene found in the B. abortus strains along with B. melitensis strains such as BM_16M and BM_Rev1 were devoid of any sequence variability. However, mutations in this gene are not associated with the development of rough phenotype (Zygmunt et al. 2009). Apart from the genes directing rough-smooth phenotype, previous studies have also identified genes associated with the oxidative stress survival of Brucella spp. Mutations in the genes coding for glutathione S-transferase family protein, NAD (P) transhydrogenase subunit alpha, magnesium transporter, and MFS efflux pump confer reduced ability to B. melitensis Rev 1 vaccine strain to cope up with the oxidative stress within the host cell resulting in an attenuated phenotype (Kornspan et al. 2020). Thus, whole genome–based analysis will help us to understand the genetic mechanism associated with the altered phenotype and attenuation of the vaccine strains compared to field strains.
As a bench mark, we applied genome sequence–based strain typing methods such as whole genome sequence–based phylogeny and cgMLST scheme along with conventional MLST schemes to infer genetic relatedness among various strains. MLST is a powerful typing system, which can help to identify pathogen origin and spreading status. The current study discriminated 5, 6, and 25 genotypes as per 9, 21, and cgMLST schemes. Different genotypes have been found in standard strains. The cgMLST shows high discriminatory as compared to other typing systems. It has the ability to discriminate strains circulating within the country. Strains from Punjab and Northeastern India grouped regionally showing circulation of similar type strains in these regions. The WGP of Brucella strains indicated separate species-specific clusters for B. abortus and B. melitensis (Foster et al. 2009). Comparative genomics of ten Brucella species reported by Wattam et al. (2009) indicated four clades in phylogenetic tree and among that B. abortus and B. melitensis formed a single clade (Wattam et al. 2009). When compared to Indian field strains, the standard B. abortus and B. melitensis strains grouped independently. Isolates BM 16M and BM rev.1 formed similar outgroups based on both whole genome and core genome based phylogenetic analysis (Azam et al. 2016, Karthik et al. 2021). The specific geographical clustering of Brucella strains was demonstrated by the distinct cluster formation of Indian strains in earlier study (Sankarasubramanian et al. 2019). Similar findings and benefits of sequence-based approaches over the conventional typing system with respect to strain differentiation potential have been reported by Sankarasubramanian et al. (2015) and Abdel-Glil et al. (2022).
Conclusion
To conclude, the study carried out the genome sequence–based comparison of standard, vaccine and Indian field strains of B. abortus and B. melitensis. Both species indicated significant sharing of genes, whereas SNP divergence was higher for B. melitensis as compared to B. abortus. Genome sequence data was found valuable for identifying variations in virulence factors and differentiating closely related standard, vaccine and field strains. Further studies can be targeted for the development of vaccines with broader protection considering the genome resource informations such as genetic homology, SNPs and virulence factors present in different Brucella species and strains.
References
Abdel-Glil MY, Thomas P, Brandt C, Melzer F, Subbaiyan A, Chaudhuri P, Harmsen D, Jolley KA, Janowicz A, Garofolo G, Neubauer H (2022) Core genome multilocus sequence typing scheme for improved characterization and epidemiological surveillance of pathogenic Brucella. J Clin Microbiol 60(8):e00311–e00322
Alcock BP, Raphenya AR, Lau TT, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen ALV, Cheng AA, Liu S, Min SY (2020) CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 48(D1):D517–D525
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC genomics 12(1):1–10
Azam S, Rao SB, Jakka P, NarasimhaRao V, Bhargavi B, Gupta VK, Radhakrishnan G (2016) Genetic characterization and comparative genome analysis of Brucella melitensis Isolates from India. Int J Genom 2016:3034756. https://doi.org/10.1155/2016/3034756
Bengtsson RJ, Simpkin AJ, Pulford CV, Low R, Rasko DA, Rigden DJ, Hall N, Barry EM, Tennant SM, Baker KS (2022) Pathogenomic analyses of Shigella isolates inform factors limiting shigellosis prevention and control across LMICs. Nat Microbiol 7(2):251–261
Bertelli C, Laird MR, Williams KP, Simon Fraser University Research Computing Group, Lau BY, Hoad G, Winsor GL, Brinkman FS (2017) IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45(W1):W30–W35
Bogaards JA, Van Der Weele P, Woestenberg PJ, Van Benthem BH, King AJ (2019) Bivalent human papillomavirus (HPV) vaccine effectiveness correlates with phylogenetic distance from HPV vaccine types 16 and 18. J Infect Dis 220(7):1141–1146
Chaudhuri P, Goswami TTK, Lalsiamthara J, Kaur G, Vishnu US, Sankarasubramanian J, Gunasekaran P, Rajendhran J (2015) Draft genome sequence of the intermediate rough vaccine strain Brucella abortus S19Δ per mutant. Genome Announc 3(6):e01336–e01315
Cloeckaert A, Grayon M, Verger JM, Letesson JJ, Godfroid F (2000) Conservation of seven genes involved in the biosynthesis of the lipopolysaccharide O-side chain in Brucella spp. Res Microbiol 151(3):209–216
Crasta OR, Folkerts O, Fei Z, Mane SP, Evans C, Martino-Catt S, Bricker B, Yu G, Du L, Sobral BW (2008) Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS One 3(5):e2193
De Barsy M, Jamet A, Filopon D, Nicolas C, Laloux G, Rual JF, Muller A, Twizere JC, Nkengfac B, Vandenhaute J, Hill DE (2011) Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell Microbiol 13(7):1044–1058
El-Sayed A, Awad W (2018) Brucellosis: Evolution and expected comeback. Int J Vet Sci Med 6(sup1):S31–S35
Etemady A, Mohammdi M, Esmaelizad M, Alamian S, Vahedi F, Aghaeipour K, Behrozikhah AM, Faghihloo E, Afshar D, Firuzyar S, Rahimi A (2015) Genetic characterization of the wboA gene from the predominant biovars of Brucella isolates in Iran Electron Physician 7(6):1381
Foster JT, Beckstrom-Sternberg SM, Pearson T, Beckstrom-Sternberg JS, Chain PS, Roberto FF, Hnath J, Brettin T, Keim P (2009) Whole-genome-based phylogeny and divergence of the genus Brucella. J Bacteriol 191(8):2864–2870
Gardner SN, Slezak T, Hall BG (2015) kSNP3. 0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31(17):2877–2878
Godfroid F, Cloeckaert A, Taminiau B, Danese I, Tibor A, De Bolle X, Mertens P, Letesson JJ (2000) Genetic organisation of the lipopolysaccharide O-antigen biosynthesis region of Brucella melitensis 16M (wbk). Res Microbiol 151(8):655–668
Guzman-Verri C, Suárez-Esquivel M, Chaves Olarte E and Moreno E (2020) Brucella genomics: macro and micro evolution
Halling SM, Peterson-Burch BD, Bricker BJ, Zuerner RL, Qing Z, Li LL, Kapur V, Alt DP, Olsen SC (2005) Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J Bacteriol 187(8):2715–2726
Hurtado R, Maturrano L, Azevedo V, Aburjaile F (2020) Pathogenomics insights for understanding Pasteurella multocida adaptation. Int J Med Microbiol 310(4):151417
Huson DH, Scornavacca C (2012) Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61(6):1061–1067
Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124. https://doi.org/10.12688/wellcomeopenres.14826.1
Karthik K, Anbazhagan S, Thomas P, Ananda Chitra M, Senthilkumar TMA, Sridhar R, Dhinakar Raj G (2021) Genome sequencing and comparative genomics of Indian isolates of Brucella melitensis. Front Microbiol 12:698069
Kornspan D, Lubkovskaia R, Mathur S, Yeheskel A, Salmon-Divon M (2020) Genomic analysis of natural rough Brucella melitensis Rev. 1 vaccine strains: identification and characterization of mutations in key genes associated with bacterial lps biosynthesis and virulence. Int J Mol Sci 21(24):9341
Ledwaba MB, Glover BA, Matle I, Profiti G, Martelli PL, Casadio R, Zilli K, Janowicz A, Marotta F, Garofolo G, Van Heerden H (2021) Whole genome sequence analysis of Brucella abortus isolates from various regions of South Africa. Microorganisms 9(3):570
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49(W1):W293–W296
Liu B, Zheng D, Jin Q, Chen L, Yang J (2019) VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res 47(D1):D687–D692
Megid J, Mathias LA, Robles CA (2010) Clinical manifestations of brucellosis in domestic animals and humans. Bentham Open
Ozer EA, Allen JP, Hauser AR (2014) Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15(1):1–17
Paulsen IT, Seshadri R, Nelson KE, Eisen JA, Heidelberg JF, Read TD, Dodson RJ, Umayam L, Brinkac LM, Beanan MJ, Daugherty SC (2002) The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci 99(20):13148–13153
Pisarenko SV, Kovalev DA, Volynkina AS, Ponomarenko DG, Rusanova DV, Zharinova NV, Khachaturova AA, Tokareva LE, Khvoynova IG, Kulichenko AN (2018) Global evolution and phylogeography of Brucella melitensis strains. BMC Genomics 19(1):1–10
Sankarasubramanian J, Vishnu US, Gunasekaran P, Rajendhran J (2019) Development and evaluation of a core genome multilocus sequence typing (cgMLST) scheme for Brucella spp. Infect Genet Evol 67:38–43
Sankarasubramanian J, Vishnu US, Sridhar J, Gunasekaran P, Rajendhran J (2015) Pan-genome of Brucella species. Indian J Microbiol 55:88–101
Schurig G, Boyle S, Sriranganathan N (1995) Brucella abortus vaccine strain RB51: a brief review. Archivos de Medicina Veterinaria (Chile) 27:19-22. https://biblioteca.inia.cl/bitstream/handle/20.500.14001/38564/NR19640.pdf?sequence=1
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069
Seemann T (2016) ABRicate: mass screening of contigs for antibiotic resistance genes. GitHub, San Francisco, CA
Tan-KK TY, Chang LY (2015) Full genome SNP-based phylogenetic analysis reveals the origin and global spread of Brucella melitensis. BMC Genomics.[Internet] 16:93
Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, Gladstone RA, Lo S, Beaudoin C, Floto RA, Frost SD (2020) Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol 21:1–21
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44(W1):W232–W235
Uelze L, Grützke J, Borowiak M, Hammerl JA, Juraschek K, Deneke C, Tausch SH, Malorny B (2020) Typing methods based on whole genome sequencing data. One Health Outlook 2:1–19
Van Straten M, Bardenstein S, Keningswald G, Banai M (2016) Brucella abortus S19 vaccine protects dairy cattle against natural infection with Brucella melitensis. Vaccine 34(48):5837–5839
Wang A, Chen Q, Wang L, Madson D, Harmon K, Gauger P, Zhang J, Li G (2019) Recombination between vaccine and field strains of porcine reproductive and respiratory syndrome virus. Emerg Infect Dis 25(12):2335
Wattam AR, Williams KP, Snyder EE, Almeida NF Jr, Shukla M, Dickerman AW, Crasta OR, Kenyon R, Lu J, Shallom JM, Yoo H (2009) Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J Bacteriol 191(11):3569–3579
Yang X, Skyberg JA, Cao L, Clapp B, Thornburg T, Pascual DW (2013) Progress in Brucella vaccine development. Front Biol 8:60–77
Zhou Z, Alikhan NF, Sergeant MJ, Luhmann N, Vaz C, Francisco AP, Carriço JA, Achtman M (2018) GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res 28(9):1395–1404
Zygmunt MS, Blasco JM, Letesson JJ, Cloeckaert A, Moriyón I (2009) DNA polymorphism analysis of Brucella lipopolysaccharide genes reveals marked differences in O-polysaccharide biosynthetic genes between smooth and rough Brucella species and novel species-specific markers. BMC Microbiol 9(1):1–13
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conception and design of study: Prasad Thomas, Anbazhagan S, Pallab Chaudhuri. Acquisition of data: Himani K. M, Karthikeyan R. Analysis and/or interpretation of data: Anbazhagan S, Jonathan Lalsiamthara. Drafting the manuscript: Anbazhagan S, Lakshmi Prakasan, Sonu S. Nair, Dinesh M, Abhishek, Ramachandra S.G, Chaturvedi V.K, Prasad Thomas.
Corresponding authors
Ethics declarations
Ethical approval
Animal experiments were not conducted for this research so ethical approval is not required.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
Figure S1: Phylogenetic tree based on the SNPs (GIF 15 kb)
ESM 2
Figure S2: Virulence factors presence or absence and sequence identity (JPG 832 kb)
ESM 3
Table S1: Metadata and assembly accession number of Brucella isolates involved in the study. Table S2: Details of phages and genomic islands predicted for Brucella species. Table S3: COGs annotations of the accessory genes predicted in the B. abortus genomes. Table S4: COGs annotations of the accessory genes predicted in the B. melitensis genomes. Table S5: Details of SNPs, (Non Synonymous and Synonymous) identified among 44 strains (XLSX 302 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
Cite this article
Anbazhagan, S., Himani, K., Karthikeyan, R. et al. Comparative genomics of Brucella abortus and Brucella melitensis unravels the gene sharing, virulence factors and SNP diversity among the standard, vaccine and field strains. Int Microbiol 27, 101–111 (2024). https://doi.org/10.1007/s10123-023-00374-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10123-023-00374-w