
Abstract
Soil salinity is regarded as severe environmental stress that can change the composition of rhizosphere soil bacterial community and import a plethora of harms to crop plants. However, relatively little is known about the relationship between salt stress and root microbial communities in groundnuts. The goal of this study was to assess the effect of salt stress on groundnut growth performance and rhizosphere microbial community structure. Statistical analysis exhibited that salt stress indeed affected groundnut growth and pod yield. Further taxonomic analysis showed that the bacterial community predominantly consisted of phyla Proteobacteria, Actinobacteria, Saccharibacteria, Chloroflexi, Acidobacteria, and Cyanobacteria. Among these bacteria, numbers of Cyanobacteria and Acidobacteria mainly increased, while that of Actinobacteria and Chloroflexi decreased after salt treatment via taxonomic and qPCR analysis. Moreover, Sphingomonas and Microcoleus as the predominant genera in salt-treated rhizosphere soils might enhance salt tolerance as plant growth-promoting rhizobacteria. Metagenomic profiling showed that series of sequences related to signaling transduction, posttranslational modification, and chaperones were enriched in the salt-treated soils, which may have implications for plant survival and salt tolerance. These data will help us better understand the symbiotic relationship between the dominant microbial community and groundnuts and form the foundation for further improvement of salt tolerance of groundnuts via modification of soil microbial community.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peanut or groundnut (Arachis hypogaea L.), an important oilseed crop cultivated worldwide, serves as a major source of vegetable oil and livestock fodder in many countries (Wang et al. 2018). As sessile lifestyle, groundnuts face a multitude of environmental stresses, including high salinity, drought, high or low temperatures, heavy metals, and microbial infections (Xu et al. 2018). Among the emerging problems in agricultural sector, high salinity is regarded as the most severe environmental stress that occurs concurrently in most cases (Zhu 2001). Salt stress can induce osmotic stress and ion toxicity, which causes growth termination and plant death, leading to considerable yield loss or even total yield failure (Boudsocq and Lauriere 2005; Munns and Tester 2008; Suzuki et al. 2012; Xu et al. 2019). Groundnut is more salt stress tolerant species than other related species, and in recent years, saline-alkali soil has been tried for the cultivation of groundnuts (Chakraborty et al. 2016; Wan et al. 2014). However, extreme conditions in saline-alkali soil still severely affect groundnut’s growth and productivity. Therefore, exploring salt stress response mechanism and further enhancing groundnut salt tolerance has become a major breeding goal in groundnut industry and saline-alkali land utilization project.
During millions of years’ evolution, plants have evolved complex morphological, physiological, and molecular strategies for salt tolerance to avoid or withstand salt stress, such as the accumulation of compatible solutes, activation of antioxidant enzymes, and regulation of terminal Na+ determinants (such as Salt Overly Sensitive 1, High-affinity K+ transporter 1, Na+/H+ antiporter) (Damodharan et al. 2018; Deinlein et al. 2014; Mickelbart et al. 2015). In addition to the well-reported salt tolerance mechanism, the root external environment has also been reported to influence salt tolerance mechanisms in plants (Ullah et al. 2018; Ullah et al. 2017).
Roots are primary organ of plants for absorbing nutrients and water from soil (Lareen et al. 2016). On the other hand, roots can also exude various chemicals and nutrients into the nearby narrow region of soil, namely rhizosphere, and attract a variety of microbes, such as bacteria, fungi, algae, and protozoa living with plant roots (Mendes et al. 2013). Biotic and abiotic stresses can alter rhizosphere microbial community structures due to microbes can sense some signal molecules from plants and allow some microbial population to increase or decrease (Naylor et al. 2017; Ullah et al. 2018). On the contrary, rhizosphere-associated microbes are active to possess diverse metabolic capabilities and exert different kinds of positive effects on plant growth and adaptation to biotic and abiotic stresses (Bai et al. 2015; Dennis et al. 2010). Over the past decade, various studies have emerged implicating members of the plant growth-promoting rhizobacteria (PGPRs) to be beneficial to stress tolerance through diverse mechanisms such as provide a buffer zone for plants against stresses, produce various plant growth-promoting hormones, or induce systemic resistance for plants (Evelin et al. 2009; Geng et al. 2018; Yang et al. 2009). In addition, microbes also alter the pH, soil structure, nutrient mineralization, and soil fertility, and these changes may also partially have implications for plant survival and stress response (Finkel et al. 2017). Thus, rhizosphere-associated microbes exert a vital role in enhancing plants stress tolerance. It is necessary to further study the colonization and propagation of microbial community to find and select the beneficial microbes for salt tolerance.
Studying the effect of salt stress on rhizosphere microbial composition and their correlation with crop performance is therefore crucial for understanding the salt stress tolerance mechanism. In this study, we investigated the effects of salt stress on groundnut growth and pod yield and the associated rhizosphere bacterial diversity using statistical analysis and 16S rRNA gene sequencing, respectively. Through a variety of analyses, we aimed to assess and provide new insight into the influence of salt stress on the composition of the bacterial community in the groundnut rhizosphere.
Materials and methods
Plant materials and salt treatment
Groundnuts (Huayu25, salt-resistant cultivars) were cultivated in the greenhouse at Laixi experimental station, China (120.53 °E, 36.86 °N). In order to characterize the bacterial community in field conditions and further perform salt stress treatment, fine groundnuts were grown in a transparent acrylic tank (36 cm in diameter and 26 cm tall) with tiny holes in the bottom containing the same weight of topsoil. The topsoil was dug from a groundnut field of the Laixi experimental station. Then the soil was dried and further sieved with a 1 cm sieve, and 18 kg soil was added to each tank in the greenhouse conditions. The physiochemical properties of soil were examined before being added to the tank: pH 7.7, EC 0.26 ds/m, organic content 13.23 g kg−1, total nitrogen 1.70 g kg−1, available phosphorous 11.7 mg kg−1, and available potassium 103.2 mg kg−1. For the salt-treated soil group, the required amount of NaCl was added into soil and stirred well to blend to attain to 2.00 g kg−1 salt concentration. Four full groundnut seeds were planted in each tank, and watered every other day to keep the soil water content at 85% of field capacity until the harvest time (at 85% of field capacity, water in the pot was kept in the capillary pores of the soil and soil salt could not be lost with the water; thus, the soil salt content was 2.00 g kg−1 during the whole growth period of groundnut) under the conditions of 28 °C and approximately 16/8-h light/dark photoperiod (Dai et al. 2019). Plants and soil samples were collected and stored in liquid nitrogen at maturity.
Growth and yield parameters of groundnut
Six representative plant samples were obtained from each treatment at maturity. Samples were preserved after being separated into leaves, stems, roots, and pods. All samples were killed by heating to 105 °C for 30 min, then dried to a constant weight at 80 °C, and weighed separately. In addition, representative plants were sampled from each treatment to record the number of pods per plant. And 100 pod weight and 100 seed weight of each treatment were also recorded.
The sample collection of rhizosphere compartments for 16S rRNA gene sequencing
Rhizosphere soils were collected as the previous study (Bulgarelli et al. 2012; Dai et al. 2019). Rhizosphere soil samples were constituted of root surface soil and soil around the roots. Soil around the roots was obtained by shaking and root surface soil was extracted by using PBS buffer according to the previous study (Geng et al. 2018). Root sections were cut off from salt and control groundnuts by sterile scissors and then placed in 50 mL of sterile centrifuge tube containing 40 mL PBS buffer (0.25 g of NaH2PO4.H2O, 0.66 g of Na2HPO4.7H2O, 8 mL Silwet L-77, pH 7.0). Then the centrifuge tubes were centrifuged at a high speed to remove the root surface soil and then filtered through a 100-mm mesh cell strainer to remove plant debris. The filtrate was centrifuged at 5000g for 15 min to form a pellet and 1 mL PBS buffer was added to the centrifuge tube to re-suspend the pellet and obtain the root surface microbes. Then the samples were frozen in liquid nitrogen at − 80 °C. Three biological replicates were performed for each treatment and a total of 6 soil groups were collected. The genomic DNA was extracted from rhizosphere soils by using PowerSoil® DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA) as the previous study (Dai et al. 2019).
16S rRNA gene sequencing and high-throughput sequencing
Extractive DNA quality and concentrations were checked by 0.8% agarose gel electrophoresis and ultraviolet spectrophotometry (Ullah et al. 2018). High-quality DNA samples were used for bacterial 16S rRNA gene amplification. 16S rRNA gene sequencing was conducted by Beijing SinoGenoMax (Beijing, China) using the Illumina HiSeq2500 platform. The specific primers 341F (forward primer, 5′-CCTACGGGNGGCWGCAG-30) and 805R (reverse primer, 5′-GACTACNVGGGTATCTAATCC-3′) with barcode were used for bacterial 16S rRNA tags (V3 and V4 region) amplification (Dai et al. 2019). PCR amplification was carried out with 2 × Phanta Max Master Mix (P515, Vazyme, Nanjing, China) and PCR products were purified with Tiangen Gel Extraction Kit (Tiangen, Beijing, China). The sequencing libraries were produced by using TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, USA) following manufacturer’s instructions. Finally, the library was sequenced on an Illumina HiSeq2500 platform and 250 bp/300 bp paired-end reads were generated. Reads were submitted to NCBI SRA under accession SUB5091314 and bioproject PRJNA517287.
OTU cluster and species annotation
Paired-end reads were merged using FLASH (version 1.2.7), which can merge paired-end reads when at least some of the reads overlap the reads generated from the opposite end of the same DNA fragment. Raw tags were preliminarily screened to obtain the high-quality clean tags according to the QIIME (Quantitative Insights into Microbial Ecology, v1.8.0) quality-controlled process (Caporaso et al. 2010). Clean tags were compared with the reference Gold database (http://drive5.com/uchime/uchime_download.html) using UCHIME algorithm to detect chimera sequences. The effective tags were obtained by using USEARCH (v5.2.236, http://www.drive5.com/usearch/) to identify and delete chimera sequences in denovo and reference way (Edgar et al. 2011). Operational taxonomic unit (OTU) was defined as the sequence of one or more samples based on a sequence similarity threshold set ≥ 97% and obtained by using UPARSE software package, which is roughly equal to the sequence difference among taxonomic species in genetic diversity analysis based on 16S rRNA genes (Blaxter et al. 2005). We picked a representative sequence for each OTU and used the RDP classifier to annotate taxonomic information for each representative sequence. Venn diagrams were plotted with the VennDiagram package to identify differences of bacterial communities among the different soil groups (Chen and Boutros 2011). Community composition data for each taxonomic level were clustered on the basis of abundance distribution of taxa or degree of similarity between soil groups through heat map, which was generated using R-package, gplots (version 3.3.1) algorithm (Zuo et al. 2017).
Alpha and beta diversity analysis
In-house Perl scripts were used to analyze alpha (within samples) and beta (among samples) diversity. In order to compute alpha diversity, we rarified the OTU table and calculated six indices: both Chao1 and ace indices estimate the species abundance; Sobs exhibits the numbers of OTUs in groundnut rhizosphere; Shannon and Simpson indices exhibit community diversity; coverage can reflect the sequencing depth to observe community richness. Rank abundance estimates the species evenness and rarefaction curves were generated based on these three metrics to evaluate the species richness and depth of sequence. Furthermore, the species accumulation curve is used to measure whether the sample size is adequate to estimate community richness. The rank abundance curve reflects the species abundance and evenness (Bates et al. 2013). Each OTU representative sequence is used for taxonomic identification of each sample at five classification levels (phylum, class, order, family, and genus).
Beta diversity analysis was conducted to examine the similarity or dissimilarities of the community structure among different soil samples. PCA and PCoA analyses were performed on the community composition structure at the genus level to explore the similarities or dissimilarities between two soil groups using QIIME software (Wang et al. 2012). Cluster analysis mainly used the distance to evaluate the similarity or dissimilarities among various soil samples.
Linear discriminant analysis effect size and COG analysis
Linear discriminant analysis (LDA) effect size (LEfSe) as an algorithm for high-dimensional biomarker was used for the quantitative analysis of biomarkers within different soil groups (Segata et al. 2011). LEfSe analysis was performed on http://huttenhower.sph.harvard.edu/galaxy with alpha value for the factorial Kruskal–Wallis test among classes was < 0.05 and the threshold on the logarithmic LDA score for discriminative features was > 3.0. PICRUSt10 program was used to predict functional features of bacterial community based on high-throughput sequencing data in the context of the COG database according to the previous study (Langille et al. 2013).
qPCR analysis of predominant bacterial community
qPCR was used to analyze the main bacterial phylum (α-Proteobacteria, β-Proteobacteria, Actinobacteria, Saccharibacteria, Chloroflexi, Acidobacteria, and Cyanobacteria) measured by the methods are described in previous study (Li et al. 2018). qPCR analysis was performed by ChamQ SYBR Color qPCR Master Mix (Q411, Vazyme, Nanjing, China) and Bio-Rad CFX96 (Bio-Rad, Hercules, CA, USA) with the following reaction mixture: 0.5 μL of primers (10 μM), 7.5 μL ChamQ SYBR Color qPCR Master Mix, and template DNA (20 ng of total soil DNA or plasmid DNA for standard curves) as the previous study (Yu et al. 2019). Primers are listed in Supplementary Table S6.
Statistical Analysis
All statistical analyses were conducted with the R program (v3.3.0, https://www.r-project.org/), and 999 displacement tests were performed to determine whether the differences between the salt-treated and untreated soil groups were statistically significant.
Results
Effects of salt stress on morphology and yield of groundnut
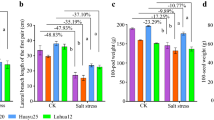
The results (Table 1) showed that the main stem height and the lateral branch length decreased under salt stress, while the number of lateral branches and leaves was not significantly different relative to controls. In addition, the pod yield of groundnut showed a downward trend in the salt-treated group. After salt stress, 100 pod weight and 100 seed weight of HY25 were reduced by 17.22% and 21.59%, respectively. The number of total pods per plant also declined after salt stress, while the difference was not significant relative to control (Table 1). Therefore, salt stress indeed suppresses growth and adversely influences pod yield of groundnut in the pot experiment.
Overall analysis of sequence data in the groundnut rhizosphere
To explore the bacterial community of the groundnut rhizosphere under normal growth and salt stress conditions, 16S rRNA gene sequencing was performed to analyze bacterial shift between normal and salt stress conditions. We combined the salt-treated root surface and rhizospheric soil to salt-treated rhizosphere (SR), and controlled root surface and rhizospheric soil to control rhizosphere (CR). Low-quality reads were filtered using the QIIME software (Quantitative Insights into Microbial Ecology, v1.8.0), and sequencing quantities of each soil sample counted were listed in Supplementary Table S1. In total, 761,664 passed quality screening for groundnut rhizosphere and most of the sequence lengths were between 440 and 460 bp (Supplementary Fig. S1a and Table S1). Operational taxonomic units (OTUs) were generated with a 97% threshold identity. A total of 3505 OTUs were found in groundnut rhizosphere soil samples, in which 3078 OTUs were shared by both the soil groups, while 210 and 217 OTUs were only present in CR and SR, respectively (Supplementary Fig. S1b).
Alpha diversity analysis of groundnut bacterial community richness and diversity
Community richness and diversity of bacterial ecosystem was examined via alpha diversity analysis. Rarefaction curve analysis showed a high sequencing depth in each groundnut rhizosphere samples (Fig. 1a). In species accumulation curves, the rate of increase of new species followed with the increase in sample size during sampling of the population, suggesting that the sequencing depth was high enough to observe community richness (Fig. 1b). Rank abundance curve showed that SR and CR had high species evenness and homogeneity (Fig. 1c). Many other indices were also performed in this study (Sobs, Shannon and Simpson, Chao1 and ace, and coverage), which can also reflect the alpha diversity of the bacterial community (Fig. 1d and Supplementary Table S2). All the results show that no significant difference in alpha diversity among the different soil samples of groundnut rhizosphere.

Alpha diversity analysis of groundnut rhizosphere bacterial community richness and diversity. a Rarefaction curve analysis showing the depth of 16S rRNA gene sequencing and the possibility of diversity. b Species accumulation curves showing the rate of increase of new species with the increase in sample size. c Rank abundance curve showing the relative species abundance and evenness. The length of polyline on horizontal axis reflects the number of operational taxonomic units (OTUs) in sample and represents the richness of bacterial community. Flatness of polyline reflects the evenness of bacterial community composition. d OTU levels via sobs index analysis showing the relative species abundance in two soil groups
Groundnut rhizosphere bacterial community structure analysis
To further analyze the bacterial community structure, abundance distributions of each sample at five levels of classification (phylum, classes, orders, families, and genus) were obtained. Average relative abundances of two soil groups were classified into 31 phyla, but only 11 phyla were found at a relative abundance of > 1% (Supplementary Table S3). Most of the bacteria were belonged to Proteobacteria, Actinobacteria, Saccharibacteria, Chloroflexi, Acidobacteria, and Cyanobacteria at the phylum level, accounted for more than 80% of bacterial sequences in both SR and CR (Fig. 2a). Saccharibacteria, Cyanobacteria, and Acidobacteria increased and dominated in SR, while the abundance of Actinobacteria, Chloroflexi, and Verrucomicrobia decreased in the salt-treated soils compared with the CR (Fig. 2a).

Bacterial community structure in groundnut rhizosphere. a The percent of bacterial community abundance at the phylum level in two soil groups in groundnut rhizosphere. The relative abundance is calculated by averaging the abundances of duplicate samples in each soil group in groundnut rhizosphere. b The percent of bacterial community abundance at the class level in two soil groups in groundnut rhizosphere. The relative abundance is calculated by averaging the abundances of duplicate samples in each soil group in groundnut rhizosphere. c The percent of bacterial community abundance at the order level in two soil groups in groundnut rhizosphere. The relative abundance is calculated by averaging the abundances of duplicate samples in each soil group in groundnut rhizosphere. d The percent of bacterial community abundance at the family level in two soil groups in groundnut rhizosphere. The relative abundance is calculated by averaging the abundances of duplicate samples in each soil group in groundnut rhizosphere. The names of “norank” and “unclassified” are all unidentified species obtained directly from database via sequence alignment
At the class level, the groundnut rhizosphere bacterial community structure consisted mainly of Actinobacteria, Alphaproteobacteria, Acidobacteria, Betaproteobacteria, Gemmatimonadetes, and Cyanobacteria (Fig. 2b and Supplementary Table S4). Compared with soil without salt treatment, Cyanobacteria increased as the dominant class in SR. However, Actinobacteria and Chloroflexia decreased in SR compared with that in CR (Fig. 2b). Sphingomonadales was the most abundant order. Sphingomonadales, SubsectionIII, and norank_p_Saccharibacteria increased in SR, whereas Micrococcales and Gaiellales demonstrated the reverse (Fig. 2c). Norank_p_Saccharibacteria, Sphingomonadaceae, Gemmatimonadaceae, Micrococcaceae, and norank_o_Acidimicrobiales were the abundant families in groundnut rhizosphere soils (Fig. 2d).
An in-depth investigation at the genus level showed that 495 taxa were classified from SR and CR, whereas most of the genera were less than 7%, implying high bacterial diversity in the two groundnut rhizosphere soil groups (Supplementary Excel S1). As shown in Fig. 3a, norank_p_Saccharibacteria, Sphingomonas, unclassified_f_Micrococcaceae, norank_o_Acidimicrobiales, unclassified_k_norank, Gemmatimonas, norank_o_Gaiellales, and Microcoleus were predominantly found in rhizosphere soil. Salt treatment increased the abundance of Sphingomonas and Microcoleus but reduced the number of unclassified_f_Micrococcaceae and norank_o_Gaiellales (Fig. 3a and Supplementary Table S5). Moreover, many sequences named as “norank” or “unclassified” were all unidentified species but were abundant in groundnut rhizosphere soils, demonstrating that the groundnut soil remained a challenging reservoir of biodiversity which needed to further study (Fig. 3a). The generic diversity between two soil groups was clearly demonstrated by Wilcoxon rank-sum test, and Sphingomonas, norank_p_Saccharibacteria, and Microcoleus were predominant genera in salt-treated groups (Fig. 3b and Supplementary Fig. S2). Thus, salt stress can quickly lead to the shift of bacterial communities and enrichment of some specific bacterial species.

Bacterial community structure and diversity in salt-treated and untreated groundnut rhizosphere. a Percent of bacterial community abundance at the genus level in two soil groups. The relative abundance is calculated by averaging the abundances of duplicate samples. b Bacterial community diversity in salt-treated and untreated groundnut rhizosphere via Wilcoxon rank-sum test bar plot. The left image represents the proportions of various genera in two soil groups. The right image represents the difference between proportions in 95% confidence intervals. The names of “norank” and “unclassified” are all unidentified species obtained directly from database via sequence alignment
Clustering analysis of rhizosphere bacterial community composition
In order to observe the similarities and dissimilarities between two soil groups, principal component analysis (PCA), principal co-ordinates analysis (PCoA), and clustering analysis were performed. PCA analysis showed distinct differences in bacterial communities between SR and CR. The first two principal components (PC1 and PC2) of PCA explained 30.4% and 20.56% of total variations, respectively (Fig. 4a). PCoA analysis also showed that the bacterial community structures of SR and CR were divergent (Fig. 4b). At the same time, analysis of similarities or dissimilarities between and within soil groups was determined, and the results revealed that there was a significant bray_curtis distance between SR and CR. However, duplicate samples within SR and CR had very little bray_curtis distance (Fig. 4c). Cluster analysis revealed that the bacterial community structure of SR and CR were diverse, whereas the duplicate samples in SR and CR were similar to other beta diversity analyses (Fig. 4d).

Beta diversity analysis. a Two-dimensional ranking of the principal component analysis (PCA) analysis. The same color points belong to the same soil group, and the same soil group points are marked by ellipses. b Multi-dimensional ranking of the principal co-ordinates analysis (PCoA) anal analysis. The same color points belong to the same soil group, and the same soil group points are marked by ellipses. c Analysis of similarities showing the similarity and dissimilarity between two soil groups. d Hierarchical clustering is clustered according to soil groups’ similarity. Branch length between soil groups represents the degree of similarity between the two soil groups. The samples belonging to the same soil group are closer to each other and the samples from different soil groups are farther apart
Phylogenetic tree analysis showed that Microcoleus and Sphingomonas were the most abundant genera in salt-treated groundnut rhizosphere (Fig. 5a and Supplementary Fig. S3). In order to check the distinct differences in bacterial community structure between the two soil groups, high and low abundance taxa of top 50 most abundant genera were distinguished via heat map. Sphingomonas and Microcoleus at the genus level were relatively abundant in the groundnut rhizosphere under salt stress conditions (Fig. 5b). Thus, all the results show that salt stress leads to the shift of bacterial communities and enrichment of Sphingomonas and Microcoleus at the genus level, which may have implications for groundnut survival and salt tolerance as PGPRs.

Taxonomic analysis through phylogenetic tree and heat map. a A phylogenetic tree showing the relationship between salt-treated and untreated soil groups. The phylogenetic tree was constructed on the basis of 16S rRNA gene sequences. Bootstrap values were obtained from a search with 1000 replicates and are shown at the nodes. Species names and proportions in two soil groups showing in the right. b The heat map visualization with hierarchical clustering of the top 50 most abundant genera was generated according to the similarity between their constituents and were arranged in a horizontal order according to the clustering results. In this figure, red represents the more abundant genera in the corresponding sample, and blue represents the less abundant genera
Specific phylotypes of groundnut rhizosphere bacterial community modulate by salt stress
Linear discriminant analysis (LDA) effect size (LEfSe) as an algorithm for high-dimensional biomarker discovery and explanation of genomic features was employed to identify specific phylotypes of groundnut rhizosphere responding to salt stress. Statistical analysis was performed from phylum to genus level in cladograms, and LDA scores of three or greater were confirmed by LEfSe (Fig. 6 and Supplementary Fig. S4). In salt-treated group, five groups of bacteria were enriched, namely Cyanobacteria (the phylum and class), SubsectionIII (the order and family, SubsectionIII and FamilyI_o__SubsectionIII), Gemmatimonadetes (from phylum to genus), Sphingomonadaceae (the family and genus, Sphingomonadaceae and Sphingomonas), and Microcoleus (genus) (Fig. 6). For example, Cyanobacteria and Microcoleus were predominant phylotypes in bacterial communities of SR but was notably low in CR in LEfSe bar (Supplementary Fig. S4). On the contrary, four groups of bacteria were detected to be enriched in CR, namely Actinobacteria (from order to genus), Micrococcales (the order Micrococcales), Rhizobiaceae (the family and genus, Rhizobiaceae and unclassified_f__Rhizobiaceae), and Gammaproteobacteria (class) (Fig. 6). Taken together, all the results indicate that Cyanobacteria as predominant phyla may be favorable bacteria for salt resistance mechanisms in groundnuts.

Cladogram showing specific phylotypes of groundnut rhizosphere responding to salt stress. Indicator bacteria with linear discriminant analysis (LDA) scores of three or greater in bacterial communities associated with soil from salt-treated and untreated soil groups. Circles indicate phylogenetic levels from phylum to genus (from the inner circle to the outer circle). The diameter of each circle is proportional to the abundance of the group. The names of “norank” and “unclassified” are all unidentified species obtained directly from database via sequence alignment
Metabolic functional features prediction of groundnut rhizosphere bacterial community
To better understand the important role of bacterial community isolated from groundnut rhizosphere, PICRUSt10 (phylogenetic investigation of communities by reconstruction of unobserved states) program was used to predict metabolic functional features of bacterial community via 16S rRNA based high-throughput sequencing data in the context of Cluster of Orthologous Groups (COG) database. Most of the metabolic functions such as amino acid transport and metabolism, carbohydrate transport and metabolism, energy production and conversion, and transcription were enriched in CR group, which implies that the bacterial metabolism tended to be vigorous in the normal groundnut rhizosphere soil samples than that in salt-treated samples (Figs. 7a, b). However, three stress-related classifications (posttranslational modification, protein turnover, chaperones; signal transduction mechanism; replication recombination and repair) were enriched in SR group (Fig. 7a). These vigorous function groups may be related to the stress response of bacterial community in the salt-treated soil group.

The bacterial functional features in salt-treated and untreated soil groups via Cluster of Orthologous Groups (COG) analysis. a Box plot showing the relative abundance and diversity of various functional groups in two soil groups. b Bar chart showing the relative abundance and diversity of functional groups in salt-treated and untreated soil groups. The relative abundance is calculated by averaging the abundances of duplicate samples. Different COG groups were displayed in different colors at the bottom
qPCR of specific bacterial groups
To further verify the 16S rRNA gene sequencing data, qPCR assay further confirmed the changes in the abundance of the main bacterial phyla (Proteobacteria, Actinobacteria, Saccharibacteria, Chloroflexi, Acidobacteria, and Cyanobacteria) in all soil samples. In addition to Saccharibacteria, the numbers of Cyanobacteria and Acidobacteria were significantly higher in salt-treated groundnut rhizosphere soils, whereas Actinobacteria and Chloroflexi were lower in these soil samples as the taxonomic analysis in Fig. 2a (Fig. 8). Most of the results were consistent with the 16S rRNA gene sequencing analysis. Taken together, all the results indicate that Cyanobacteria and Acidobacteria as predominant phyla in salt-treated soils may be favorable bacteria for salt resistance mechanisms in groundnuts.

Quantification of genes involved in major bacterial communities under different groundnut rhizosphere soils. The genes of a α-Proteobacteria, b β-Proteobacteria, c Actinobacteria, d Saccharibacteria, e Chloroflexi, f Acidobacteria, g Cyanobacteria. Error bars indicate SEM (N = 3), p < 0.05. One-way ANOVA Duncan’s test was used for statistical analysis. Statistical differences are indicated by lowercase letters and different letters represent different significance. These experiments were repeated three times
Discussion
Salinity is the main threat worldwide declining agricultural productivity. In our study, soil salinity is adversely affecting the growth and yield of groundnuts. The main stem height, lateral branch length, and pod yield showed a significant downward trend in the salt-treated group (Table 1). Therefore, it is urgently needed to ameliorate the impact of saline land on global groundnut production by some efficient approach. At present, the imperfection of groundnut genome sequence and the difficulty of genetic transformation hinder the molecular mechanism studies and genetic engineering of groundnut to some extent. Rhizosphere-associated microbes have been reported to influence salt tolerance mechanisms in plants (Ullah et al. 2018; Ullah et al. 2017). Studying the colonization of bacterial community and further selecting and inoculating the beneficial bacteria may be a practical and meaningful strategy to alleviate the impact of saline land on groundnut production to some extent. In the current work, the groundnut bacterial community diversity under salt stress and normal conditions was examined via high-throughput sequencing of 16S rRNA gene. Here, salt stress can quickly lead to the shift of bacterial community and enrichment of specific bacterial species in groundnut rhizosphere soils. The relative abundance of Cyanobacteria and Acidobacteria increased, while that of Actinobacteria and Chloroflexi decreased in salt-treated rhizosphere compared with that in rhizosphere under normal conditions via verifying by qPCR assay (Figs. 2 and 8). Cyanobacteria is crucial in improving soil environments via accumulating soil carbon and nitrogen (Bullerjahn and Post 2014). Previous study has demonstrated that Cyanobacteria can survive in saline-alkali soil (Masuda and Enrich-Prast 2016; Singh 2018). Moreover, it has been well documented that Acidobacteria show great adaptive abilities and grow well in an environment poor in nutrients or containing high concentration of salt (Foesel et al. 2014; Szymanska et al. 2018). Therefore, the higher Cyanobacteria and Acidobacteria in salt-treated rhizosphere may help groundnuts maintain necessary physiological functions under salt stress and then enhance the ability to withstand salt stress.
Taxonomic analysis showed that the bacterial community structure predominantly consisted of genera Sphingomonas, norank_p_Saccharibacteria, unclassified_f_Micrococcaceae, norank_o_Acidimicrobiales, Gemmatimonas, Microcoleus and norank_o_Gaiellales in the groundnut rhizosphere soils under normal and salt stress conditions (Fig. 3). Salt treatment increased the abundance of Sphingomonas and Microcoleus but reduced the abundance of unclassified_f_Micrococcaceae (Fig. 3). The salt-induced microbes, Sphingomonas and Microcoleus, may enhance plant growth and salt tolerance as PGPRs. Sphingomonads are often found in hostile environments due to their ability to degrade a wide range of toxic substances (Peng et al. 2008). Moreover, Sphingomonas are believed to play an important role in nitrogen fixation and denitrification in the nitrogen cycle (Xie and Yokota 2006). The role of Microcoleus in Cyanobacteria phylum in plant growth promotion under salt stress and desiccation condition have also been well documented (Masuda and Enrich-Prast 2016; Tashyreva and Elster 2015). Thus, these salt-induced rhizobacteria Sphingomonas and Microcoleus may enhance groundnut growth and improve tolerance to salt stress in some specific mechanisms, such as the improvement of the nitrogen cycle and nutrient uptake, scavenging of toxic substances as PGPRs. These await further study. The soil bacterial community might be important to plants in maintaining essential functions under salt stress. In studying the predicted function features of the bacterial community, we found that three stress-related groups (posttranslational modification, protein turnover, chaperones; signal transduction mechanism; replication recombination and repair) were predicted to be higher in salt-treated soils (Fig. 7). Some chaperones, such as heat shock proteins, are known as salt tolerance enhancers for plants (Fu et al. 2016; Guan et al. 2018). Higher signal transduction mechanism in the bacteria of salt-treated soils may confer high tolerance levels to stress and toxic compounds as PGPRs (Dai et al. 2019; Wu et al. 2018). In addition, the vigorous basic biological process, replication recombination, and repair in salt-treated rhizosphere may also contribute to improving stress tolerance for groundnuts (Dai et al. 2019). In the further study, we will propagate and inoculate moderately beneficial bacteria into the rhizosphere soil and analyze the relationship between salt stress tolerance and pod yield of groundnut and soil microbial community. Managing rhizosphere microbes and maintaining the balance of beneficial and harmful microbes in the soil are crucial to the effectiveness of cropping practices.
References
Bai Y et al (2015) Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528:364–369. https://doi.org/10.1038/nature16192
Bates ST, Clemente JC, Flores GE, Walters WA, Parfrey LW, Knight R, Fierer N (2013) Global biogeography of highly diverse protistan communities in soil. ISME J 7:652–659. https://doi.org/10.1038/ismej.2012.147
Blaxter M, Mann J, Chapman T, Thomas F, Whitton C, Floyd R, Abebe E (2005) Defining operational taxonomic units using DNA barcode data. Philos Trans R Soc Lond B Biol Sci 360:1935–1943. https://doi.org/10.1098/rstb.2005.1725
Boudsocq M, Lauriere C (2005) Osmotic signaling in plants: multiple pathways mediated by emerging kinase families. Plant Physiol 138:1185–1194. https://doi.org/10.1104/pp.105.061275
Bulgarelli D et al (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95. https://doi.org/10.1038/nature11336
Bullerjahn GS, Post AF (2014) Physiology and molecular biology of aquatic cyanobacteria. Front Microbiol 5:359. https://doi.org/10.3389/fmicb.2014.00359
Caporaso JG et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Chakraborty K, Bhaduri D, Meena HN, Kalariya K (2016) External potassium (K(+)) application improves salinity tolerance by promoting Na(+)-exclusion, K(+)-accumulation and osmotic adjustment in contrasting peanut cultivars. Plant Physiol Biochem 103:143–153. https://doi.org/10.1016/j.plaphy.2016.02.039
Chen H, Boutros PC (2011) VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. https://doi.org/10.1186/1471-2105-12-35
Dai L, Zhang G, Yu Z, Ding H, Xu Y, Zhang Z (2019) Effect of drought stress and developmental stages on microbial community structure and diversity in peanut rhizosphere soil. Int J Mol Sci 20. https://doi.org/10.3390/ijms20092265
Damodharan K, Palaniyandi SA, Le B, Suh JW, Yang SH (2018) Streptomyces sp. strain SK68, isolated from peanut rhizosphere, promotes growth and alleviates salt stress in tomato (Solanum lycopersicum cv. Micro-Tom). J Microbiol 56:753–759. https://doi.org/10.1007/s12275-018-8120-5
Deinlein U, Stephan AB, Horie T, Luo W, Xu G, Schroeder JI (2014) Plant salt-tolerance mechanisms. Trends Plant Sci 19:371–379. https://doi.org/10.1016/j.tplants.2014.02.001
Dennis PG, Miller AJ, Hirsch PR (2010) Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol Ecol 72:313–327. https://doi.org/10.1111/j.1574-6941.2010.00860.x
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. https://doi.org/10.1093/bioinformatics/btr381
Evelin H, Kapoor R, Giri B (2009) Arbuscular mycorrhizal fungi in alleviation of salt stress: a review. Ann Bot 104:1263–1280. https://doi.org/10.1093/aob/mcp251
Finkel OM, Castrillo G, Herrera Paredes S, Salas Gonzalez I, Dangl JL (2017) Understanding and exploiting plant beneficial microbes. Curr Opin Plant Biol 38:155–163. https://doi.org/10.1016/j.pbi.2017.04.018
Foesel BU et al (2014) Determinants of Acidobacteria activity inferred from the relative abundances of 16S rRNA transcripts in German grassland and forest soils. Environ Microbiol 16:658–675. https://doi.org/10.1111/1462-2920.12162
Fu C, Liu XX, Yang WW, Zhao CM, Liu J (2016) Enhanced salt tolerance in tomato plants constitutively expressing heat-shock protein in the endoplasmic reticulum. Genet Mol Res 15. https://doi.org/10.4238/gmr.15028301
Geng LL, Shao GX, Raymond B, Wang ML, Sun XX, Shu CL, Zhang J (2018) Subterranean infestation by Holotrichia parallela larvae is associated with changes in the peanut (Arachis hypogaea L.) rhizosphere microbiome. Microbiol Res 211:13–20. https://doi.org/10.1016/j.micres.2018.02.008
Guan P, Wang J, Li H, Xie C, Zhang S, Wu C, Yang G, Yan K, Huang J, Zheng C (2018) Sensitive to SALT1, an endoplasmic reticulum-localized chaperone, positively regulates salt resistance. Plant Physiol 178:1390–1405. https://doi.org/10.1104/pp.18.00840
Langille MG et al (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. https://doi.org/10.1038/nbt.2676
Lareen A, Burton F, Schafer P (2016) Plant root-microbe communication in shaping root microbiomes. Plant Mol Biol 90:575–587. https://doi.org/10.1007/s11103-015-0417-8
Li Q et al (2018) Belowground interactions impact the soil bacterial community, soil fertility, and crop yield in maize/peanut intercropping systems. Int J Mol Sci 19. https://doi.org/10.3390/ijms19020622
Masuda LS, Enrich-Prast A (2016) Benthic microalgae community response to flooding in a tropical salt flat. Braz J Biol 76:577–582. https://doi.org/10.1590/1519-6984.18314
Mendes R, Garbeva P, Raaijmakers JM (2013) The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev 37:634–663. https://doi.org/10.1111/1574-6976.12028
Mickelbart MV, Hasegawa PM, Bailey-Serres J (2015) Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nat Rev Genet 16:237–251. https://doi.org/10.1038/nrg3901
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59:651–681. https://doi.org/10.1146/annurev.arplant.59.032607.092911
Naylor D, DeGraaf S, Purdom E, Coleman-Derr D (2017) Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J 11:2691–2704. https://doi.org/10.1038/ismej.2017.118
Peng RH et al (2008) Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev 32:927–955. https://doi.org/10.1111/j.1574-6976.2008.00127.x
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. https://doi.org/10.1186/gb-2011-12-6-r60
Singh H (2018) Desiccation and radiation stress tolerance in cyanobacteria. J Basic Microbiol 58:813–826. https://doi.org/10.1002/jobm.201800216
Suzuki N, Koussevitzky S, Mittler R, Miller G (2012) ROS and redox signalling in the response of plants to abiotic stress. Plant Cell Environ 35:259–270. https://doi.org/10.1111/j.1365-3040.2011.02336.x
Szymanska S, Borruso L, Brusetti L, Hulisz P, Furtado B, Hrynkiewicz K (2018) Bacterial microbiome of root-associated endophytes of Salicornia europaea in correspondence to different levels of salinity. Environ Sci Pollut Res Int 25:25420–25431. https://doi.org/10.1007/s11356-018-2530-0
Tashyreva D, Elster J (2015) Effect of nitrogen starvation on desiccation tolerance of Arctic Microcoleus strains (cyanobacteria). Front Microbiol 6:278. https://doi.org/10.3389/fmicb.2015.00278
Ullah A, Sun H, Yang X, Zhang X (2017) Drought coping strategies in cotton: increased crop per drop. Plant Biotechnol J 15:271–284. https://doi.org/10.1111/pbi.12688
Ullah A, Akbar A, Luo Q, Khan AH, Manghwar H, Shaban M, Yang X (2018) Microbiome diversity in cotton Rhizosphere under Normal and drought conditions. Microb Ecol. https://doi.org/10.1007/s00248-018-1260-7
Wan L et al (2014) Identification of ERF genes in peanuts and functional analysis of AhERF008 and AhERF019 in abiotic stress response. Funct Integr Genomics 14:467–477. https://doi.org/10.1007/s10142-014-0381-4
Wang Y, Sheng HF, He Y, Wu JY, Jiang YX, Tam NF, Zhou HW (2012) Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl Environ Microbiol 78:8264–8271. https://doi.org/10.1128/AEM.01821-12
Wang X et al (2018) Genomic and transcriptomic analysis identified gene clusters and candidate genes for oil content in peanut (Arachis hypogaea L.) plant. Mol Biol Report 36:518–529. https://doi.org/10.1007/s11105-018-1088-9
Wu L et al (2018) Comparative metagenomic analysis of rhizosphere microbial community composition and functional potentials under rehmannia glutinosa Consecutive Monoculture. Int J Mol Sci 19. https://doi.org/10.3390/ijms19082394
Xie CH, Yokota A (2006) Sphingomonas azotifigens sp. nov., a nitrogen-fixing bacterium isolated from the roots of Oryza sativa. Int J Syst Evol Microbiol 56:889–893. https://doi.org/10.1099/ijs.0.64056-0
Xu Y, Yu Z, Zhang D, Huang J, Wu C, Yang G, Yan K, Zhang S, Zheng C (2018) CYSTM, a novel non-secreted cysteine-rich peptide family, involved in environmental stresses in Arabidopsis thaliana. Plant Cell Physiol 59:423–438. https://doi.org/10.1093/pcp/pcx202
Xu Y et al (2019) CYSTM3 negatively regulates salt stress tolerance in Arabidopsis. Plant Mol Biol 99:395–406. https://doi.org/10.1007/s11103-019-00825-x
Yang J, Kloepper JW, Ryu CM (2009) Rhizosphere bacteria help plants tolerate abiotic stress. Trends Plant Sci 14:1–4. https://doi.org/10.1016/j.tplants.2008.10.004
Yu Z et al (2019) CEPR2 phosphorylates and accelerates the degradation of PYR/PYLs in Arabidopsis. J Exp Bot 70:5457–5469. https://doi.org/10.1093/jxb/erz302
Zhu JK (2001) Plant salt tolerance. Trends Plant Sci 6:66–71
Zuo Y, Xie W, Pang Y, Li T, Li Q, Li Y (2017) Bacterial community composition in the gut content of Lampetra japonica revealed by 16S rRNA gene pyrosequencing. PLoS One 12:e0188919. https://doi.org/10.1371/journal.pone.0188919
Funding
The work was financially supported by the National Key R&D Program of China (2018YFD1000906) and the Agricultural Scientific and the Technological Innovation Project of Shandong Academy of Agricultural Sciences (CXGC2018E21).
Author information
Authors and Affiliations
Contributions
Z.Z. and L.D. conceived and designed the study; Y.X. and G.Z. performed research, analyzed the data, and made the figures; Y.X. wrote the original article; H.D. and D.C. contributed new methods and provided important suggestions; Z.Z. and Y.X. supervised and complemented the writing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 677 kb).
Rights and permissions
About this article

Cite this article
Xu, Y., Zhang, G., Ding, H. et al. Influence of salt stress on the rhizosphere soil bacterial community structure and growth performance of groundnut (Arachis hypogaea L.). Int Microbiol 23, 453–465 (2020). https://doi.org/10.1007/s10123-020-00118-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10123-020-00118-0