
Abstract
The rapidly growing mycobacterium Mycobacterium abscessus is a clinically important organism causing pulmonary and skin diseases. The M. abscessus complex is comprised of three subspecies: M. abscessus subsp. abscessus, M. abscessus subsp. massiliense, and M. abscessus subsp. bolletii. Here, we aimed to develop a Cas12a/sgRNA-based nucleic acid detection platform to identify M. abscessus species and subspecies. By designing specific sgRNA probes targeting rpoB and erm(41), we demonstrated that M. abscessus could be differentiated from other major mycobacterial species and identified at the subspecies level. Using this platform, a total of 38 clinical M. abscessus isolates were identified, 18 as M. abscessus subsp. abscessus and 20 as M. abscessus subsp. massiliense. We concluded that the Cas12a/sgRNA-based nucleic acid detection platform provides an easy-to-use, quick, and cost-effective approach for identification of M. abscessus species and subspecies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-tuberculous Mycobacteria (NTM) have been recognized as important human pathogens that cause concerns for human health [1,2,3]. Among the NTM species, the Mycobacterium abscessus complex belonging to the subgroup of rapidly growing Mycobacteria (RGM) are important human pathogens, accounting for the majority of NTM lung infections among cystic fibrosis patients [4]. Pulmonary infections caused by the Mycobacterium. abscessus complex often last for many years and they tend to become refractory diseases due to their resistance to most antibiotics used in the clinic [5, 6]. The M. abscessus complex can be divided into three subspecies, M. abscessus subsp. abscessus, M. abscessus subsp. bolletii, and M. abscessus subsp. massiliense [7, 8]. It has been reported that the three M. abscessus subspecies have different antibiotic resistance profiles and may cause different clinical symptoms [5, 9, 10]. Therefore, rapid and accurate identification of M. abscessus and the precise subspecies is critical to the treatment and control of M. abscessus infection.
Accurate identification of M. abscessus at the subspecies level remains complicated. Multilocus sequence typing (MLST) is an ubiquitous method to define isolates by using the sequences of several housekeeping genes [11]. However, it is tedious and expensive. Some studies have reported that improved matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) could be employed to rapidly and cost-effectively identify both the M. abscessus complex and its subspecies in clinical laboratories [12,13,14,15,16]. This method is cheap, rapid, and powerful, but requires sophisticated machinery. Recently, the commercially available kit GenoType NTM-DR (Hain Lifescience, Nehren, Germany) has been frequently used not only for subspecies differentiation of M. abscessus but also for determination of molecular resistance [17, 18]. However, this method also needs expensive equipment. Actually, whole-genome sequencing (WGS) is the most accurate and reliable approach for the identification of bacterial species, but it is expensive and time-consuming. With the development of next generation sequencing (NGS) technology, it may be practical to employ whole-genome sequencing for identification of bacterial species in clinical diagnosis in the future. However, currently there is an urgent need to develop a rapid and accurate method to identify both the M. abscessus complex and its subspecies.
Here, we aimed to develop an easy-to-use, fast, and cost-effective method to identify M. abscessus species and subspecies. Recently, the advanced CRISPR/Cas12a-based nucleic acid detection platform has shown dramatically high sensitivity in nucleic acid detection with single base resolution [19,20,21]. We therefore reasoned it can be developed to identify the M. abscessus complex and subspecies. In the present study, by designing specific sgRNA probes targeting rpoB or erm(41), the Cas12a/sgRNA-based nucleic acid detection platform could distinguish the M. abscessus complex from other major mycobacterial species and identify the subspecies of M. abscessus.
Methods and materials
Bacteria collection and DNA extraction
In the present study, clinical mycobacterial isolates were obtained from the Shenzhen Third People’s Hospital affiliated with the Southern University of Science and Technology. These isolates were previously identified using gene chip (YanengBIO, Shenzhen, China) and further confirmed by sequencing the rpoB gene using previously published primers [22]. The reference mycobacterial strains were obtained from the China Center for Disease Control and Prevention. All strains were cultured in Middlebrook 7H9 liquid medium. DNA was extracted using a simple boiling method [23].
PCR and sequencing of rpoB and hsp65 genes
The rpoB and hsp65 genes were amplified from clinical isolates DNA using previously published primers [11]. PCR was performed using Ex-Taq DNA Polymerase (TaKaRa, Dalian, China) in a 50 μL reaction volume as follows: one cycle at 94 °C for 3 min, then 30 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s, followed by one cycle at 72 °C for 5 min, cooling at 4 °C. PCR products were examined using 1% agarose gel electrophoresis and then directly sequenced. DNA sequences from the type strains of M. abscessus subsp. abscessus (CIP 104536T), M. abscessus subsp. bolletii (CIP 108541T), and M. abscessus subsp. massiliense (CIP 108297T) were retrieved from the GenBank database.
Phylogenetic analyses
The sequences of the rpoB and hsp65 genes were aligned with the ClustalW algorithm using MEGA 6.0 software. Phylogenetic trees based on the sequences of the rpoB and hsp65 genes were constructed by the neighbor-joining method with 1000 bootstrap replications.
Design and preparation of sgRNA probes targeting the rpoB gene
The rpoB and erm(41) genes were used as the targets to identify the M. abscessus complex and its subspecies in this study. rpoB sequences of M. tuberculosis H37Rv and the most common NTM species (M. abscessus subsp. abscessus ATCC 19977, M. abscessus subsp. bolletii CIP 108541T, M. abscessus subsp. massiliense CIP 108297T, M. intracellulare ATCC 13950, M. avium ATCC 25291, M. kansasii ATCC 12478, M. gordonae ATCC 14470, and M. fortuitum ATCC 6841) were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/). Multiple alignments of the sequences were performed using DNAMAN 8 software. We firstly designed M. abscessus-specific sgRNA1 probes based on the divergence of the rpoB gene to distinguish the M. abscessus complex from other mycobacterial species. Subsequently, two sgRNA probes (sgRNA2 and sgRNA3) were designed based on the diversity sequence of rpoB within the M. abscessus subspecies. Then a sgRNA4 probe targeting the C-terminal deletion of erm(41) was designed to distinguish M. abscessus subsp. massiliense from M. abscessus subsp. abscessus and M. abscessus subsp. bolletii. Primer pairs for amplification of the targets were designed and are listed in supplementary Table S1. The DNA templates for the sgRNA transcription were synthesized by Songon Biotech (Shanghai, China). All sgRNA probes were prepared by in vitro transcription using a T7 High Yield RNA Transcription Kit (Vazyme, Nanjing, China) according to the manufacturer’s instructions. The transcribed RNA was purified using VAHTS RNA Clean Beads (Vazyme, Nanjing, China) and quantified with NanoDrop2000. All of the oligonucleotides are listed in Table S1.
PCR and Cas12a trans-cleavage assay
Target genes of rpoB and erm(41) were amplified from the strains by PCR with the primers listed in supplementary Table S1. The PCR procedures were performed using the following steps, one cycle at 94 °C for 3 min, then 30 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s, followed by one cycle at 72 °C for 5 min, cooling at 4 °C. The PCR products were examined using 1% agarose gel electrophoresis. The concentrations of the PCR products were measured through a Qubit dsDNA HS Assay Kit (ThermoFisher Scientific, Massachusetts, USA).
Recombinant Francisella novicida Cas12a (FnCas12a) protein purchased from Tolo Biotech (Shanghai, China) was used for the Cas12a trans-cleavage assay. Cas12a trans-cleavage assays were performed according to Li’s previously published description [19]. In brief, an FnCas12a trans-cleavage assay was conducted in a reaction buffer consisting of 0.5 pmol FnCas12a, 100 nM purified sgRNA probes, target DNA (1 μl unpurified PCR products), 500 nM collateral ssDNA (quenched fluorescent DNA reporter), and 10 U RNase inhibitor (TaKaRa, Dalian, China) in a 20 μl volume at 37 °C for 1 h. The reaction was stopped by adding 2 μl of 0.25 M EDTA. Fluorescence emission was excited at 535 nm and detected at 560 nm using a Varioskan Flash (ThermoFisher Scientific, Massachusetts, USA), and reactions with no target DNA were taken as the background.
Results
Schematic overview of the Cas12a/sgRNA-based nucleic acid detection platform
The principle of the development of the FnCas12a/sgRNA-based nucleic acid detection platform for identification of bacteria is illustrated in Fig. 1. To improve the sensitivity of this platform, PCR or other nucleic acid amplification methods are employed to enrich target DNA. Specific sgRNA probes direct Cas12a to recognize target DNA that is complementary to the sgRNA, which thereby triggers Cas12a to trans-cleave the reporter ssDNA, allowing it to fluoresce.

Illustration of the Cas12a/sgRNA-based nucleic acid detection platform. The target DNA is specifically amplified from the extracted DNA of the bacteria by PCR or other methods. Specific sgRNA probes are designed to match the target DNA. The PCR product is mixed with the FnCas12a/sgRNA probe/ssDNA-FQ (quenched fluorescent single strand DNA reporter). Once the sgRNA probe matches the target, FnCas12a will trans-cleave the quenched fluorescent ssDNA reporter, leading to fluorescence
Design of the sgRNA probes for identification of the M. abscessus complex and subspecies
In order to identify the M. abscessus complex, a M. abscessus-specific sgRNA1 probe was designed based on the divergence of the rpoB genes among the major Mycobacterium species (Fig. 2a). Alignment of the rpoB sequences from type strains of M. abscessus ATCC 19977, M. abscessus subsp. bolletii CIP 108541T, and M. abscessus subsp. massiliense CIP 108297T revealed different sequences that could be potential loci to distinguish M. abscessus subsp. abscessus from M. abscessus subsp. massiliense and M. abscessus subsp. bolletii (Fig. 2b). To determine whether these different regions were robust among M. abscessus subspecies, we downloaded a number of rpoB sequences from strains of M. abscessus, M. abscessus subsp. massiliense, and M. abscessus subsp. bolletii from NCBI GenBank. Multiple alignment of these downloaded sequences confirmed different regions among the three M. abscessus subspecies (Fig. 2c). Subsequently, two specific sgRNAs (sgRNA2 and sgRNA3) were designed to target the different regions to differentiate M. abscessus subsp. abscessus from M. abscessus subsp. massiliense and M. abscessus subsp. bolletii (Fig. 2b).

Design of the sgRNA probes for identification of M. abscessus (a) and subspecies (b), (c) based on rpoB sequences. a To design a specific sgRNA probe for identification of the M. abscessus complex, rpoB sequences from the type strains of major mycobacterial species were downloaded. b and c To find the potential difference of regions for identification of M. abscessus subspecies, rpoB sequences were downloaded from many strains of M. abscessus, including type strains of M. abscessus subsp. abscessus (CIP 104536T), M. abscessus subsp. bolletii (CIP 108541T), and M. abscessus subsp. massiliense (CIP 108297T). Multiple sequence alignment was performed using DNAMAN 8 software. Red boxes represent sgRNA probe targeted regions. Green boxes represent the FnCas12a recognition “TTN” motif. Red stars represent the differences among subspecies of M. abscessus
However, these probes were not reliable for differentiating M. abscessus subsp. massiliense from M. abscessus subsp. bolletii due to the extremely low rpoB divergence between these two subspecies. The M. abscessus subsp. massiliense erm(41) gene has been reported to contain a large C-terminal deletion [9, 24]. Therefore, a specific sgRNA4 probe targeting the C-terminal deletion region was designed to distinguish M. abscessus subsp. massiliense from M. abscessus subsp. abscessus and M. abscessus subsp. bolletii.
Determination of the feasibility of the Cas12a/sgRNA-based nucleic acid detection platform for identification of M. abscessus and M. abscessus subspecies
Firstly, we determined whether the designed sgRNA1 probe is able to discriminate the M. abscessus complex from other mycobacterial species by using reference mycobacterial strains. PCR was employed to amplify the target rpoB gene. As shown in Fig. 3a, according to the results of the fluorescence intensity, M. abscessus strains showed strong fluorescence intensity compared with control, while other reference mycobacterium strains showed no significant increase in fluorescence relative to the control. Subsequently, a number of identified clinical isolates were used to further test the specificity of the sgRNA1 probe in a blinded manner. All of the clinical M. abscessus strains were correctly identified without any cross-reaction, indicating the high specificity of the sgRNA1 probe (Table 1).

Application of the FnCas12a/sgRNA-based nucleic acid detection platform for identification of M. abscessus species and subspecies. a Determination of the specificity of the designed sgRNA1 probes for identification of M. abscessus using the major mycobacterial reference strains and an E. coli negative control. b Identification of subspecies of M. abscessus among the 38 clinical M. abscessus isolates using sgRNA2 and sgRNA3 probes. Arabic numerals represent clinical M. abscessus isolates (two-tailed Student’s t test; ****p < 0.0001; bars represent the mean ± SEM; n = three technical replicates)
To determine the feasibility of sgRNA2 and sgRNA3 in discrimination of M. abscessus subsp. abscessus from M. abscessus subsp. massiliense and M. abscessus subsp. bolletii, 38 confirmed clinical M. abscessus strains were subjected to this assay. As shown in Fig. 3b, 18 were identified as M. abscessus subsp. abscessus and 20 strains were identified as M. abscessus subsp bolletii or M. abscessus subsp. massiliense based on the fluorescence intensity, which is consistent with the results of phylogenetic trees based on the sequences of rpoB or hsp65 (Fig. 4).

Construction of phylogenetic trees based on the partial rpoB and hsp65 genes of 38 clinical M. abscessus isolates. Both trees were constructed using the neighbor-joining method in the MEGA 6.0 program. The bootstrap values were calculated from 1000 replications. Arabic numerals represent clinical M. abscessus isolates
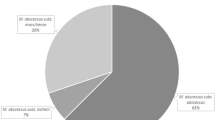
Since the sgRNA2 and sgRNA3 probes were both not able to discriminate M. abscessus subsp. massiliense from M. abscessus subsp. bolletii, erm(41) PCR was applied to distinguish these two closely related subspecies. Both M. abscessus subsp. abscessus and M. abscessus subsp. bolletii subspecies are expected to produce 700 bp PCR products while the M. abscessus subsp. massiliense subspecies produces an approximately 350 bp PCR product. The agarose gel results showed that 18 strains displayed a full-size product (approximately 700 bp) while 20 strains showed a truncated product (approximately 350 bp) (Fig. 5), indicating 18 strains belong to M. abscessus subsp. abscessus or M. abscessus subsp. bolletii subspecies and 20 strains belong to M. abscessus subsp. massiliense. Meanwhile, we obtained the same results as the erm(41) PCR using the Cas12a-based detection platform with the sgRNA4 probe (data not shown). Taken together with the results of the Cas12/sgRNA cleavage assay, 18 and 20 strains, respectively, were identified as M. abscessus subsp. abscessus and M. abscessus subsp. massiliense among these 38 clinical M. abscessus isolates.

The PCR results of erm(41) genes from the 38 clinical M. abscessus isolates. Arabic numerals represent clinical M. abscessus isolates. The M. abscessus subsp. abscessus or M. abscessus subsp. bolletii erm(41) PCR products are fully amplified to 700 bp in size. The M. abscessus subsp. massiliense amplicons are amplified to 350 bp in size. Among the 38 clinical M. abscessus strains, 18 produced 700 bp PCR products and 20 strains produced 350 bp PCR products
Discussion
The M. abscessus complex is one of the most important groups of rapidly growing mycobacteria that cause pulmonary infections. Although the classification of its subspecies remains controversial, the M. abscessus complex is currently divided into three subspecies: M. abscessus subsp. abscessus, M. abscessus subsp. bolletii, and M. abscessus subsp. massiliense. However, accurately and quickly identifying the M. abscessus complex and its subspecies remains a real challenge [25].
In the present study, we described the application of the Cas12a/sgRNA-based nucleic acid detection platform for differentiation of M. abscessus strains at the subspecies level. Here, the rpoB gene was selected as the candidate gene due to its higher discriminatory power in the identification of mycobacterial species [26]. By designing a M. abscessus species-specific sgRNA1 probe, the M. abscessus complex was easily and quickly differentiated from other major mycobacterial species. A total of 38 clinical isolates were correctly identified as M. abscessus at the species level using the Cas12a/sgRNA-based nucleic acid detection platform, indicating the high accuracy of this platform in the detection of M. abscessus species. Several studies have also reported the use of the rpoB gene to identify M. abscessus at the subspecies level, although intergroup lateral transfers of rpoB exist among the M. abscessus subspecies [7, 27, 28]. Interestingly, multiple sequence alignment revealed distinct regions that could be exploited to distinguish M. abscessus subsp. abscessus from M. abscessus subsp. bolletii and M. abscessus subsp. massiliense. By designing subspecies-specific sgRNA probes (sgRNA2 and sgRNA3) targeting the different regions, we identified 18 and 20 strains as M. abscessus subsp. abscessus and M. abscessus subsp. bolletii or M. abscessus subsp. massiliense, respectively. These results are consistent with the phylogenetic trees based on rpoB and hsp65 gene sequences, suggesting the feasibility of identification of subspecies of M. abscessus merely by relying on specific detection of these distinct regions. Alternatively, we can simply design two TaqMan probes based on the distinct regions to differentiate M. abscessus subsp. abscessus from M. abscessus subsp. bolletii and M. abscessus subsp. massiliense.
Since the divergence of the rpoB sequence between the type strains of M. abscessus subsp. massiliense and M. abscessus subsp. bolletii is below 3%, it is not reliable for distinguishing M. abscessus subsp. massiliense from M. abscessus subsp. bolletii. Subsequently, erm(41) PCR as a second step was performed to distinguish M. abscessus subsp. massiliense from M. abscessus subsp. bolletii, because M. abscessus subsp. massiliense suffers from a large C-terminal deletion [9, 24]. Combining all of these results, we finally accurately identified 18 M. abscessus subsp. abscessus and 20 M. abscessus subsp. massiliense specimens among the 38 clinical isolates, and no M. abscessus subsp. bolletii strains were identified. These results suggest that M. abscessus subsp. massiliense and M. abscessus subsp. abscessus are the two primary M. abscessus subspecies in the south of China.
There are also several limitations to this study. For example, the clinical isolates are so limited that we didn’t identify any cases of M. abscessus subsp. bolletii. This assay also requires two separate PCR steps, one for amplification of rpoB and the other for amplification of erm(41). In the future, we aim to develop a Multiplex-PCR to amplify both targets in one PCR reaction, which will reduce the number of experimental steps and simplify the assay.
In conclusion, we developed an assay that could easily and quickly identify M. abscessus species. The assay consists of two stepwise detections. First, the M. abscessus complex is discriminated from other mycobacterial species using the M. abscessus-specific sgRNA1 probe and the Cas12a/sgRNA-based nucleic acid detection platform. Subsequently, subspecies of M. abscessus are identified using the subspecies sgRNA probes (sgRNA2 and sgRNA3) and erm(41) PCR. Since the Cas12a/sgRNA-based nucleic acid detection platform doesn’t require any expensive instruments, it will be popular in clinical microbiology laboratories and can be used to accelerate early diagnosis and treatment of mycobacterial infections.
References
Donohue MJ, Wymer L (2016) Increasing prevalence rate of nontuberculous mycobacteria infections in five states, 2008–2013. Annals of the American Thoracic Society 13:AnnalsATS.201605-353OC
Donohue MJ (2018) Increasing nontuberculous mycobacteria reporting rates and species diversity identified in clinical laboratory reports. BMC Infect Dis 18:163
Yu X, Liu P, Liu G, Zhao L, Hu Y, Wei G, Luo J, Huang H (2016) The prevalence of non-tuberculous mycobacterial infections in mainland China: systematic review and meta-analysis. J Infect 73:558
Harris KA, Kenna DTD, Cornelis B, Hartley JC, Turton JF, Paul A, Dixon GLJ (2012) Molecular fingerprinting of Mycobacterium abscessus strains in a cohort of pediatric cystic fibrosis patients. J Clin Microbiol 50:1758–1761
Won-Jung K, Kyeongman J, Nam Yong L, Bum-Joon K, Yoon-Hoh K, Seung-Heon L, Young Kil P, Ki KC, Sung Jae S, Huitt GA (2011) Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med 183:405
Rebecca G, Byrd TF (2008) Differential antibiotic susceptibility of Mycobacterium abscessus variants in biofilms and macrophages compared to that of planktonic bacteria. Antimicrob Agents Chemother 52:2019–2026
Toidi A, Pierre B, Didier R, Michel D (2006) rpoB gene sequence-based characterization of emerging non-tuberculous mycobacteria with descriptions of Mycobacterium bolletii sp. nov., Mycobacterium phocaicum sp. nov. and Mycobacterium aubagnense sp. nov. Int J Syst Evol Microbiol 56:133
Toidi A, Martine RG, Gilbert G, Marie-José G, Bernard LS, Didier R, Michel D (2004) Amoebal coculture of “Mycobacterium massiliense” sp. nov. from the sputum of a patient with hemoptoic pneumonia. J Clin Microbiol 42:5493
Hee-Youn K, Byoung Jun K, Yoonwon K, Yeo-Jun Y, Jeong Hwan S, Bum-Joon K, Yoon-Hoh K (2010) Mycobacterium massiliense is differentiated from Mycobacterium abscessus and Mycobacterium bolletii by erythromycin ribosome methyltransferase gene (erm) and clarithromycin susceptibility patterns. Microbiol Immunol 54:347–353
Toshiyuki H, Yasushi A, Atsuyuki K, Hideaki N, Kazunari T, Takashi F, Syuichi Y, Eriko S, Toshihiko K, Akira K (2012) Clinical and microbiological differences between Mycobacterium abscessus and Mycobacterium massiliense lung diseases. J Clin Microbiol 50:3556–3561
Macheras E, Konjek J, Roux AL, Thiberge JM, Bastian S, Leão SC, Palaci M, Sivadon-Tardy V, Gutierrez C, Richter E (2014) Multilocus sequence typing scheme for the Mycobacterium abscessus complex. Res Microbiol 165:82–90
Theofano P, Pincus DH, Dorothy G, Melissa J, Josephine B, Julian P, R Andres F, Peter G. (2015) Mycobacterium abscessus complex identification with matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 53:2355–2358
Mellmann A, Cloud J, Maier T, Keckevoet U, Ramminger I, Iwen P, Dunn J, Hall G, Wilson D, Lasala P (2008) Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J Clin Microbiol 46:1946–1954
Fangous MS, Mougari F, Gouriou S, Calvez E, Raskine L, Cambau E, Payan C, Héry-Arnaud G (2014) Classification algorithm for subspecies identification within the Mycobacterium abscessus species, based on matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 52:3362–3369
Tseng SP, Teng SH, Lee PS, Wang CF, Yu JS, Lu PL (2013) Rapid identification of M. abscessus and M. massiliense by MALDI-TOF mass spectrometry with a comparison to sequencing methods and antimicrobial susceptibility patterns. Future Microbiol 8:1381–1389
Kehrmann J, Wessel S, Murali R, Hampel A, Bange FC, Buer J, Mosel F (2016) Principal component analysis of MALDI TOF MS mass spectra separates M. abscessus (sensu stricto) from M. massiliense isolates. BMC Microbiol 16:24
Kehrmann J, Kurt N, Rueger K, Bange FC, Buer J (2016) GenoType NTM-DR for identifying Mycobacterium abscessus subspecies and determining molecular resistance. J Clin Microbiol 54:JCM.00147–JCM.00116
Mougari F, Loiseau J, Veziris N, Bernard C, Bercot B, Sougakoff W, Jarlier V, Raskine L, Cambau E (2017) Evaluation of the new GenoType NTM-DR kit for the molecular detection of antimicrobial resistance in non-tuberculous mycobacteria. J Antimicrob Chemother 72
Li SY, Cheng QX, Wang JM, Li XY, Zhang ZL, Gao S, Cao RB, Zhao GP, Wang J (2018) CRISPR-Cas12a-assisted nucleic acid detection. Cell Discovery 4:20
Li SY, Cheng QX, Liu JK, Nie XQ, Zhao GP, Wang J (2018) CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res
Chen JS, Ma E, Harrington LB, Costa MD, Doudna JA. 2018. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360:eaar6245
Toidi A, Philippe C, Michel D (2003) rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol 41:5699–5708
Mikaeili F, Kia EB, Sharbatkhori M, Sharifdini M, Jalalizand N, Heidari Z, Zarei Z, Stensvold CR, Mirhendi H (2013) Comparison of six simple methods for extracting ribosomal and mitochondrial DNA from Toxocara and Toxascaris nematodes. Exp Parasitol 134:155–159
Blauwendraat C, Dixon GLJ, Hartley JC, Foweraker J, Harris KA (2012) The use of a two-gene sequencing approach to accurately distinguish between the species within the Mycobacterium abscessus complex and Mycobacterium chelonae. Eur J Clin Microbiol Infect Dis 31:1847–1853
Leung JM, Olivier KN (2013) Nontuberculous mycobacteria: the changing epidemiology and treatment challenges in cystic fibrosis. Curr Opin Pulm Med 19:662–669
De Zwaan R, van Ingen J, van Soolingen D (2014) Utility of rpoB gene sequencing for identification of nontuberculous mycobacteria in the Netherlands. J Clin Microbiol 52:2544–2551
Nie W, Duan H, Huang H, Lu Y, Bi D, Chu N (2014) Species identification of Mycobacterium abscessus subsp. abscessus and Mycobacterium abscessus subsp. bolletii using rpoB and hsp65, and susceptibility testing to eight antibiotics. Int J Infect Dis 25:170–174
Hee-Youn K, Yoonwon K, Yeo-Jun Y, Geun PC, Nam Yong L, Tae Sun S, Bum-Joon K, Yoon-Hoh K (2008) Proportions of Mycobacterium massiliense and Mycobacterium bolletii strains among Korean Mycobacterium chelonae-Mycobacterium abscessus group isolates. J Clin Microbiol 46:3384
Funding
This work was supported by the China Postdoctoral Science Foundation (No. 2019 M653108), the Natural Science Foundation of China (No. 81873958), the National Science and Technology Major Project for Control and Prevention of Major Infectious Diseases of China (No. 2017ZX10103004), the State Key Laboratory of Respiratory Diseases Open Project (No. SKLRD-OP-201919), the Key Laboratory of Tropical Disease Control of the Ministry of Education Open Project (No.2019kfkt01, 2019kfkt04), the Shenzhen Scientific and Technological Foundation (JCYJ20180306170510974, JCYJ20180228162453330, and JCYJ20180228162511084), and the Sanming Project of Medicine in Shenzhen (No. SZSM201911009).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This study was approved by the Institutional Review Board of the Shenzhen Third People’s Hospital.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Xiao, G., Zhang, S., Liang, Z. et al. Identification of Mycobacterium abscessus species and subspecies using the Cas12a/sgRNA-based nucleic acid detection platform. Eur J Clin Microbiol Infect Dis 39, 551–558 (2020). https://doi.org/10.1007/s10096-019-03757-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-019-03757-y