
Abstract
This study aimed to use whole-genome sequencing to determine virulence and antimicrobial resistance genes in K. pneumoniae isolated from patients in a tertiary care hospital in Kilimanjaro. K. pneumoniae isolates from patients attending Kilimanjaro Christian Medical Centre between August 2013 and August 2015 were fully genome-sequenced and analysed locally. Sequence analysis was done for identification of virulence and AMR genes. Plasmid and multi-locus sequence typing and capsular or capsular (K) typing were performed and phylogeny was done to ascertain K. pneumoniae relatedness. Stata 13 (College Station, TX, 77845, USA) was used to determine Cohen’s kappa coefficient of agreement between the phenotypically tested and sequence-predicted resistance. A total of 16 (47.1%) sequence types (STs) and 10 (29.4%) K types were identified in 30 (88.2%) and 17 (50.0%) of all analysed isolates, respectively. K. pneumoniae ST17 were 6 (17.6%). The commonest determinants were blaCTX-M-15 in 16 (47.1%) isolates, blaSHV in 30 (88.2%), blaOXA-1 in 8 (23.5%) and blaTEM-1 in 18 (52.9%) isolates. Resistance genes for aminoglycosides were detected in 21 (61.8%) isolates, fluoroquinolones in 13 (38.2%) and quinolones 34 (100%). Ceftazidime and ceftriaxone showed the strongest agreement between phenotype- and sequence-based resistance results: 93.8%, kappa = 0.87 and p = 0.0002. Yersiniabactin determinant was detected in 12 (35.3%) of K. pneumoniae. The proportion of AMR and virulence determinants detected in K. pneumoniae is alarming. WGS-based diagnostic approach has showed promising potentials in clinical microbiology, hospital outbreak source tracing virulence and AMR detection at KCMC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Klebsiella pneumoniae is a major cause of hospital acquired infections including pneumonia, bloodstream and new-borns infection [1,2,3,4]. Treating infections caused by K. pneumoniae and other Enterobacteriaceae has been challenging worldwide due to emergence and rapid spread of resistant strains [5,6,7,8,9]. Initiatives towards monitoring of AMR should nonetheless go hand-to-hand with identification strategies of hypervirulent K. pneumoniae (hvKP) strains. Because roles played by virulence factors (VFs) in hvKP like yersiniabactin [10], colibactin [11], aerobactin and salmochelin [12] in enhancing severity of infections and increase their survival is significant. The global dissemination of hvKP clones [13,14,15] do pose a serious public health threat, thus underscoring a necessity to characterise VFs in K. pneumoniae. Treatment failures and possibly many deaths could now be attributed to K. pneumoniae infections [16,17,18]. Reports have generally described that treating bacterial infections in low- and middle-income countries (LMICs) is adding more burden on the already disease-burdened communities [19,20,21] both psychosocially and economically. In some countries, it is believed that even carbapenems will no longer treat more than a half of K. pneumoniae infections [22]. Having a bacterial infection will soon mean death if measures against irrational prescription and misuse of antibiotics are not taken seriously [23,24,25]. Antibiotics have been over prescribed and over-consumed especially in LMICs [26, 27]. These practices have been implicated to be fuelling up drug resistance selection pressure [28, 29].
In LMICs like Tanzania, lack of efficient clinical laboratory diagnostic systems is one of the factors often leading to empirical treatment. We previously reported the huge potential for whole-genome sequencing (WGS) to improve clinical diagnostics and infection control at a tertiary hospital in Tanzania where clinical laboratories lack access to molecular-based methodologies for regular typing of bacterial isolates [30, 31]. In this report, we used WGS to determine molecular relatedness, antimicrobial resistance genes, virulence genes and plasmids diversity in K. pneumoniae isolates from patients at KCMC, which is a tertiary care hospital in Kilimanjaro, Tanzania.
Materials and methods
Study design, participants and specimen collection
A hospital-based prospective cross-sectional study was conducted at KCMC hospital from 2013 to 2015. Part of the study’s methods has been described in details by Kumburu et al. [32]. Geographically, KCMC is located in Moshi municipality in Kilimanjaro and it is one of the biggest referral hospitals in Tanzania. It serves as a zonal referral hospital for a catchment area of around 15 million people. The hospital has a bed capacity of 650 with approximately 500 outpatients seeking medical services daily. This study was granted ethical approval by the KCMUCo Research Ethics Committee and the National Institute for Medical Research in Tanzania. A written informed consent was obtained from each participant or from parents or guardians of children before enrolment into the study. A convenient sampling method was used to recruit the study participants. It included participants suspected to have bacterial infection and admitted in medical and surgical wards. Specimens collected for bacterial culture included sputum, wound or pus swab and stool samples. Bacteria culture, isolation and identification were performed following in-house standard operating procedures as well as the Clinical and Laboratory Standards Institute (CLSI) guidelines. Sequentially, all K. pneumoniae isolates recovered over the study period were included for whole-genome sequencing and analysis. Over a 2-year period, 590 samples were collected without apriori knowledge of the infecting agent. A total of 377 bacterial strains were isolated, and whole genome sequenced. A number of isolates from this collection were randomly selected for antimicrobial susceptibility testing. A total of 34 K. pneumoniae collected sequentially were included in this study; amongst which, 16 K. pneumoniae isolates had phenotype-based antimicrobial susceptibility results.
Genomic DNA isolation, whole genome sequencing and analysis
For all K. pneumoniae isolates, genomic DNA (gDNA) was purified and its concentration was determined using the Easy-DNA Extraction Kit (Invitrogen®) and the Qubit dsDNA Assay Kit (Invitrogen®), respectively. The gDNA library preparation was performed following Nextera® XT DNA Sample Preparation Guide [33]. In brief, each gDNA was tagmented (tagged and fragmented) by the Nextera® XT transposome. The transposome simultaneously fragments the input DNA and adds adapter sequences to the fragment ends. Then, a limited-cycle PCR amplification followed, whereby indexes required for cluster formation were added to each DNA piece. Then, each gDNA library was normalised to ensure equal representation during sequencing. Equal volumes of the normalised library were combined, diluted in hybridization buffer and heat denatured prior to sequencing on the Illumina MiSeq platform (Illumina Inc.). The sequencer output was analysed using the standard WGS pipeline at KCRI, which is based on local implementations of the bioinformatics services available at https://cge.cbs.dtu.dk/services/. Quality control of the reads was performed using FastQC 0.11.4 [34]. De novo assembly was performed with SPAdes 3.11.1 [35], and quality assessed using QUAST 4.5 [36]. For this article’s purpose, the analyses included resistance gene identification using ResFinder 2.1 [37], multi-locus sequence typing (MLST) determination using MLST 1.8 [38], plasmid and plasmid MLST determination using PlasmidFinder 1.3 and pMLST 1.4 [39] and virulence gene determination using VirulenceFinder 1.4 [40]. Phylogeny reconstruction was done using CSI Phylogeny [41] (reference NTUH-K2044). The 34 assembled K. pneumoniae genomes of the present study have been submitted to the European Nucleotide Archive (ENA) with project accession number PRJEB26616. Stata 13 (College Station, TX, 77845, USA) was used to determine Cohen’s kappa coefficient of agreement between the phenotype- and whole-genome sequence-based antimicrobial resistance results.
Results
Study participants and Klebsiella isolates
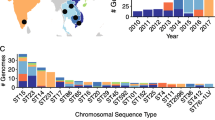
A total of 34 K. pneumoniae isolates were recovered: 9 (26.5%) in 2013, 17 (50.0%) in 2014 and 8 (23.5%) in 2015. Out of 34 K. pneumoniae, 25 (73.5%) isolates were from wound or pus swabs, 5 (14.7%) from sputum, 3 (8.8%) from stool and 1 (2.9%) from throat swab. Sixteen (47.1%) of K. pneumoniae were isolated from surgical wards, 3 (8.8%) from surgical ICU, 12 (35.3%) were isolated from patients admitted in medical wards, 1 (2.9%) from medical ICU and 2 (5.9%) were isolated from outpatients. A total of 11 (32.4%) K. pneumoniae were isolated from participants with infected wounds. The proportion of K. pneumoniae from participants with cough was 6 (17.6%), burn 6 (17.6%), wounds 6 (17.6%) and diabetes 6 (17.6%) (Table 1).
MLST and capsular (K) typing
A total of 16 (47.1%) STs were identified in 30 (88.2%) of the analysed isolates whilst the remaining isolates could not be typed (unknown STs, 4 (11.8%)). A total of 6 (17.6%) were K. pneumoniae ST17, of which 4 were recovered from patients in surgical and 2 in medical wards. A total of 4 (10.8%) were K. pneumoniae ST392, of which 2 were recovered from patients in medical and 2 in surgical wards. Three (8.8%) were K. pneumoniae ST348 and all were recovered from patients in surgical wards. K. pneumoniae ST15, ST25, ST299 and ST1562 each was observed in 2 (5.4%) of the isolates (Table 2). A total of 10 (29.4%) different K types were identified including K2, K7, K10, K19, K23, K28, K34, K41, K60 and K80 whilst isolates with unknown K types were 17 (50.0%). All 4 K. pneumoniae ST392 were of unknown K types, whereas 5 out of 6 K. pneumoniae ST17 were unknown K types (Table 2).
Beta-lactam-resistance determinants
All isolates were carrying at least one beta-lactam-resistance determinant. The commonest detected determinants were blaCTX-M-15 in 16 (47.1%) isolates, blaSHV in 30 (88.2%), blaOXA-1 in 8 (23.5%) and blaTEM-1 in 18 (52.9%) isolates. A variable population of blaSHV genes was found, whereby blaSHV-11 was found in 15 (44.1%) and blaSHV-1 in 7 (20.6%) isolates. Other infrequently blaSHV-detected genes included blaSHV-12, blaSHV-28, blaSHV-61, blaSHV-83, blaSHV-99 and blaSHV-133. At least three beta-lactam- resistance genes were found in 15 (44.1%) isolates. Carriages of four beta-lactam-resistance genes, namely blaCTX-M-15, blaSHV-11, blaOXA-1 and blaTEM-1B, were frequently observed in K. pneumoniae ST392 isolates (Table 2).
Aminoglycoside, fluoroquinolone and quinolone determinants
Almost all isolates were carrying at least one resistance determinant for aminoglycosides in 21 (61.8%) isolates, fluoroquinolones in 13 (38.2%) and quinolones 34 (100%). Fluoroquinolones gene, aac(6’)Ib-cr, was identified in 13 (38.2%) isolates. Determinants for aminoglycoside resistance included aph(3″)-Ib and aph(6)-Id in 16 (47.1%) and 17 (50.0%) isolates, respectively. Other infrequent determinants for aminoglycosides were aac(3)-IIa, aac(3)-IId, aph(3′)-Ia, aadA16, aadA1, aadA5, aadA24 and aadB (Table 3). The quinolone resistance determinants identified were oqxAB in all 34 (100%) and qnrB in 11 (32.4%) isolates. The variants for qnrB gene included qnrB1, qnrB6, qnrB49 and qnrB66.
Fosfomycin, macrolide and phenicol
The determinants FosA and mph(A) for fosfomycin and macrolide resistance were identified in 34 (100%) and 4 (11.8%) isolates, respectively. Several gene families for phenicol resistance were detected: catA2 in 10 (29.4%), catA1 in 2 (5.9%), catB4 in 8 (10.8%) and cmlA1 in 2 (5.9%) isolates.
Rifampicin, sulphonamide tetracycline and trimethoprim
Seven (20.6%) isolates were carrying arr-3, a rifampicin resistance gene. Sulphonamide resistance determinants, sul1 and sul2, were detected in 12 (35.3%) and in 22 (64.7%) isolates, respectively. Two tetracycline resistance genes detected were tet(A) in 7 (20.6%) and tet(D) in 4 (11.8%) isolates. Trimethoprim resistance genes were identified in 23 (67.6%) isolates. A total of 11 (32.4%) isolates were carrying dfrA14 and 5 (13.5%) carrying dfrA27. Other infrequently observed trimethoprim resistance genes detected included dfrA(1/5/7/15/17/25/30) and dfrG (Table 3).
Comparison of phenotype- and whole-genome sequence-based antimicrobial resistance
Agreement between phenotype- and whole-genome sequence-based antimicrobial resistance was done for 16 out of 34 K. pneumoniae isolates (Table 4). On average, agreement across all antibiotics tested was 77.4%. Overall, the phenotypically determined resistance was higher than the whole-genome sequence-based resistance. Nevertheless, all antibiotics but ampicillin showed substantial (61–80%) or strong agreement (81–100%) between phenotype- and sequence-based resistance results. Ampicillin showed moderate agreement: 56.3%, kappa = 0.13 and p = 0.1508. The sequence-based analysis predicted that resistance to ampicillin was in 19 (55.9%) isolates, whereas phenotypic testing revealed 15 (93.8%) of isolates to be resistant. Ceftazidime and ceftriaxone showed the strongest agreement: 93.8%, kappa = 0.87 and p = 0.0002. Sequence-based analysis predicted resistance to both ceftazidime and ceftriaxone in 16 (47.1%) of isolates, whereas 6 (37.5%) of isolates were resistant phenotypically.
Virulence determinants in K. pneumoniae
Further, we analysed virulence determinants in all K. pneumoniae strains. Yersiniabactin was detected in a significant proportion, in 12 (35.3%) of isolates out of which, all (n = 3) K. pneumoniae ST348 were carrying yersiniabactin genes. The ferric uptake operon system (kfuABC) was found in 10 (29.4%) of isolates. Different from the rest, isolate 315 (K. pneumoniae ST2042) was carrying genes coding for aerobactin, salmochelin and yersiniabactin but lacked the ferric uptake operon system (Table 5).
Plasmid multi-locus sequence typing
Plasmid analysis revealed a high diversity of incompatibility groups (Inc) or plasmid replicons (Table 6). The most frequent replicon was IncFIB(K) that was found in 30 (88.2%) K. pneumoniae isolates. Other IncFI members included IncFIB(pKPHS1), IncFIA(HI1), IncFIB(pECLA) and IncFIB(pENTAS01). Another prevalent replicon IncR was identified in 18 (52.9%) of K. pneumoniae isolates. The IncFII was another frequently detected replicons. In this group, IncFII(K) was found in 14 (41.2%), and IncFII in 7 (20.6%). Others were IncFII(pCRY), IncFII(pECLA) and IncFII(Y). Plasmid replicons that were infrequently identified include IncHI1B, IncHI2, IncHI2A and IncN3. A high diversity of plasmid multi-locus sequence types (pMLSTs) was identified (Table 6). IncF[F-:A16:B-] was detected in 5 (14.7%) of K. pneumoniae isolates, IncF[F-:A-:B10 detected in 4 (11.8%) of K. pneumoniae isolates and 9 (26.5%) and 5 (14.7%) of K. pneumoniae isolates had unknown pMLSTs belonging to IncF[Unknown ST] and IncHI1[Unknown ST], respectively. Other pMLSTs identified are shown in Table 6.
Phylogenetic analysis
For epidemiological tracking of nosocomial infections, SNP difference was calculated to show how closely or distantly related the isolates are. The observed minimum and maximum number of SNP difference between K. pneumoniae isolates were 57 and 42,893, respectively (Supplemental Table 1). The minimum SNP difference was observed between isolates 134 and 131E (both ST392), showing a possibility of nosocomial infections. The maximum SNP difference was observed between K. pneumoniae isolates ADE ST37 and 16 ST231, showing that it is unlikely these isolates closely related. The tree topology showed two isolates (79C ST 297 and 302 with undetermined ST) segregating very distinctly from others. The heatmap showed clear patterns of high beta-lactam, aminoglycoside and quinolone resistance gene proportions spreading almost universally across all isolates. Furthermore, an apparent pattern was observed indicating an inverse correlation between yersiniabactin genes across the isolates (Fig. 1).

Phylogenetic analysis of 31 K. pneumoniae isolates showing STs, resistance and virulence genes. The heatmap shows the frequency of AMR and virulence genes present in an isolate. The stronger the red colour is, the higher the number of genes identified across antibiotic classes is. The green colour stands for isolates from which a virulence gene was present
Discussion
The present study used whole-genome sequence-based approach in characterising clinical K. pneumoniae isolated from hospitalised patients at KCMC hospital in Moshi, Tanzania. All isolates were analysed to determine (1) K. pneumoniae subtypes and molecular relatedness for establishing existence of nosocomial transmissions or outbreaks, (2) virulence and antibiotic resistance determinants and (3) types of plasmids. The present study reveals high diversity of K. pneumoniae in the hospital. The observed K. pneumoniae diversity is plausibly attributed to the fact that specimens were collected from a diverse population as this is a consultant hospital that is serving the northern, eastern and central zones of Tanzania. Nonetheless through MLST, the majority of K. pneumoniae that were clonally related were actually isolates from patients admitted to the same wards. For instance, K. pneumoniae ST17 with number 17, 29 and 320 were from surgical wards. Also, instance K. pneumoniae ST17 with number 41 and 284 were from medical wards. Although few numbers of strains were identified within distinct ST groups (clusters), this may be an indication of nosocomial transmissions or outbreaks within the hospital. Similar to the present report, polyclonal existence of K. pneumoniae with predominance of K. pneumoniae ST17 in hospital settings was reported in the Netherlands by Souverein et al. [42]. Identification of K. pneumoniae clones particularly ST17 and ST348 within surgical wards in the present study compares with the findings in Norway [43] and Mwanza, Tanzania [6]. In both reports, it was shown that K. pneumoniae ST17 and ST348 strains were the likely causes of neonatal sepsis and outbreaks in neonatal ICU. Given the superiority of WGS over classical approaches in microbial identification, typing and tracing of outbreak sources [44,45,46], the possibility that there were sporadic nosocomial transmissions of K. pneumoniae in this hospital becomes highly likely.
Our data further suggests that K. pneumoniae circulating in the hospital are carrying high proportions of antimicrobial resistance determinants. These findings are in line with findings of study done in Kenya on K. pneumoniae isolates from stool [47]. This study identifies multiple carriages of resistance determinants including those for beta-lactams: blaSHV, blaCTX-M-15 and blaTEM-1. Despite the fact that we noted blaSHV being the most prevalent determinant, Tellevik et al. [48] and Mshana et al. [6] had earlier reported blaCTX-M-15 as the most prevalent determinant in Dar es Salaam and Mwanza, respectively. On average, a strong agreement was observed between phenotype- and sequence-based resistance to all antibiotics tested, findings that are consistent with the previous report on E. coli that was conducted in the same settings [49]. Nonetheless, ampicillin revealed the lowest but moderate agreement between the two methods. The phenotypically determined resistance to ampicillin was higher than sequence-based resistance. Plausibly, the observed difference could be due to the fact that WGS analysis uses only known resistance genes and it is also true that not all genes involved in resistance mechanisms have been included in these databases.
The observed multiple carriage of beta-lactam- resistance determinants in this study, particularly amongst K. pneumoniae ST17 and ST392 isolates, might substantially be a reason for their persistence in this hospital as also noted elsewhere [6]. Apart from being prevalent in this study, K. pneumoniae ST392 appeared to carry multi-resistance determinants. Findings are similar to those reported in a hospital-based study at an Italian hospital [50], which showed that K. pneumoniae ST392 strain might become very aggressive. Although we could not identify a single resistance gene for carbapenem in the present study’s isolates, K. pneumoniae ST15 and ST348 have been reported in Portugal to be the cause of KPC outbreaks [51]. The identification of this aggressive ST348 strain in this hospital should at least signal for the emergence and spread of MDR bacteria and that no sooner than later common infections caused by K. pneumoniae and other bacteria will become untreatable.
The co-carriage of aminoglycoside, fluoroquinolone and quinolone resistance determinants was very common in almost all K. pneumoniae. The fluoroquinolone, aminoglycoside and quinolone resistance genes: aac(6’)Ib-cr, oqxA and oqxB, appeared to be associated with the carriage of blaCTX-M-15 and other beta-lactam- resistance determinants, findings that are consistent with a report [52] on K. pneumoniae strains from urban settings in Barcelona.
The abundance and variability of resistance determinants to sulphonamides, tetracyclines, fluoroquinolones and trimethoprims was found to be high, findings that are in line with those by Taitt et al. [47]. In the current study, we further found a relatively higher proportion of K. pneumoniae carrying arr-3 for rifampicin resistance than the proportion documented by Taitt et al. [47]. Uncontrolled and excessive use of first- and second-line antibiotics for both clinical and veterinary purposes is a plausible explanation to the emergence and spread of these determinants in Enterobacteriaceae [53].
We further observed a significant proportion of yersiniabactin, colibactin, aerobactin and salmochelin in these K. pneumoniae strains. However, yersiniabactin was the most prevalent VF and it has been associated with K. pneumoniae infection rather than carriage, findings consistent with Holt et al. [54]. Based on the molecular characteristics proposed by Li et al. [55] for a hypervirulent K. pneumoniae, isolate 315 (K. pneumoniae ST2042) was the likely candidate. It was carrying genes coding for regulators of mucoid phenotype (rmpA), aerobactin (iucABCD and iutA), salmochelin (iroBCDN) and yersiniabactin (ybt, fyuA and irp1/2) but lacking the ferric uptake operon (kfuABC). Interestingly, we observe that this strain 315 (K. pneumoniae ST2042) with high virulence potential has low AMR. Apart from fosA and oqxAB, which it shares with all other isolates, its only beta-lactam gene is blaSHV-99, which notably none of the other isolates possess. This combination of high virulence and low AMR has been observed elsewhere [54, 56]. Amongst our isolates with many AMR genes, virulence determinants tend to be reduced.
Further analyses revealed that the most frequent plasmid replicon identified was IncF (I/II). Other replicons that were infrequently identified included IncHI1 and IncN3. The plasmids carried by K. pneumoniae appeared to be highly diverse. However, the IncFII plasmids seem to be common and correlated with the observed carriage of blaCTX-M-15, similar to a Moroccan study [57] that identified IncFII plasmid as a carrier of blaCTX-M-15 amongst K. pneumoniae ST466 strains. For instance K. pneumoniae ST15 appeared to carry plasmid ST IncF[K9:A13:B-], and K. pneumoniae ST17 was carrying plasmid ST IncF[K2*:A13:B-]. Plasmid ST IncF[K8:A-:B-] was identified in K. pneumoniae ST348 and ST231 and plasmid ST IncF[K7:A-:B-] was found in K. pneumoniae ST392 and ST29.
We acknowledge the presence of several limitations to this study. First, due to small numbers of isolates, the study lacked epidemiological analysis that might have shown correlation between AMR and virulence genes with patients’ demographics (gender, age) and clinical characteristics including admission outcomes, antibiotics use, hospitalisation history and comorbidities. Second, phenotype-based resistance results were available for small numbers of bacterial isolates; this may have impacted on the agreement between phenotype- and sequence-predicted resistance results. Third, WGS analysis relied on resistance and virulence databases that at time of analysis might comprise of known genes and not all genes involved in resistance or virulence mechanisms have been documented or included in those databases. Further, the existence of genes encoding different resistance and virulence factors do not necessarily indicate gene activity in the isolates. There is therefore a need for future genomics studies to focus on quantifying expression levels of genes encoding different resistance and virulence factors.
Conclusions
In this study, the amount of antimicrobial resistance and virulence determinants detected in K. pneumoniae is alarming. Besides its application for research purposes, in resource-limited settings, WGS-based diagnostic approach has showed promising potentials in clinical microbiology, hospital outbreak source tracing, virulence and AMR detection. Having been implemented successfully in Kilimanjaro, WGS can be used as a surveillance tool for infectious agent and AMR detection nationwide. It has the potential of accelerating informed decisions in formulation of pragmatic antimicrobial stewardships, and other infection prevention and control initiatives.
References
Christopher A, Mshana SE, Kidenya BR, Hokororo A, Morona D (2013) Bacteremia and resistant gram-negative pathogens among under-fives in Tanzania. Ital J Pediatr 39:1–8
Onken A, Said AK, Jørstad M, Jenum PA, Blomberg B (2015) Prevalence and antimicrobial resistance of microbes causing bloodstream infections in unguja, Zanzibar. PLoS One 10:e0145632. https://doi.org/10.1371/journal.pone.0145632
Pourakbari B, Sadr A, Ashtiani MTH, Mamishi S, Dehghani M, Mahmoudi S et al (2012) Five-year evaluation of the antimicrobial susceptibility patterns of bacteria causing bloodstream infections in Iran. J Infect Dev Ctries 6:120–125
Mpogoro FJ, Mshana SE, Mirambo MM, Kidenya BR, Gumodoka B, Imirzalioglu C (2014) Incidence and predictors of surgical site infections following caesarean sections at Bugando medical Centre, Mwanza, Tanzania. Antimicrob Resist Infect Control 3:25. https://doi.org/10.1186/2047-2994-3-25
Sonda T, Kumburu H, van Zwetselaar M, Alifrangis M, Lund O, Kibiki G et al (2016) Meta-analysis of proportion estimates of Extended-Spectrum-Beta-Lactamase-producing Enterobacteriaceae in East Africa hospitals. Antimicrob Resist Infect Control 5:1–9. https://doi.org/10.1186/s13756-016-0117-4
Mshana SE, Hain T, Domann E, Lyamuya EF, Chakraborty T, Imirzalioglu C (2013) Predominance of Klebsiella pneumoniae ST14 carrying CTX-M-15 causing neonatal sepsis in Tanzania. BMC Infect Dis 13:466. https://doi.org/10.1186/1471-2334-13-466
Souli M, Galani I, Antoniadou A, Papadomichelakis E, Poulakou G, Panagea T et al (2010) An outbreak of infection due to β-lactamase Klebsiella pneumoniae Carbapenemase 2–Producing K. Pneumoniae in a Greek University hospital: molecular characterization, epidemiology, and outcomes. Clin Infect Dis 50:364–373. https://doi.org/10.1086/649865
Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M et al (2013) Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis 13:785–796. https://doi.org/10.1016/S1473-3099(13)70190-7
Ahmed M, Moremi N, Mirambo MM, Hokororo A, Mushi MF, Seni J et al (2015) Multi-resistant gram negative enteric bacteria causing urinary tract infection among malnourished underfives admitted at a tertiary hospital, northwestern, Tanzania. Ital J Pediatr 41:44. https://doi.org/10.1186/s13052-015-0151-5
Bachman MA, Oyler JE, Burns SH, Caza M, Lépine F, Dozois CM et al (2011) Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect Immun 79:3309–3316. https://doi.org/10.1128/IAI.05114-11
Lu M-C, Chen Y-T, Chiang M-K, Wang Y-C, Hsiao P-Y, Huang Y-J et al (2017) Colibactin contributes to the hypervirulence of pks+ K1 CC23 Klebsiella pneumoniae in Mouse Meningitis Infections. Front Cell Infect Microbiol 7:103. https://doi.org/10.3389/fcimb.2017.00103
Russo TA, Olson R, MacDonald U, Beanan J, Davidson BA (2015) Aerobactin, but not yersiniabactin, salmochelin, or enterobactin, enables the growth/survival of hypervirulent (hypermucoviscous) Klebsiella pneumoniae ex vivo and in vivo. Infect Immun 83:3325–3333. https://doi.org/10.1128/IAI.00430-15
Pomakova DK, Hsiao CB, Beanan JM, Olson R, MacDonald U, Keynan Y et al (2012) Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: An emerging and under-recognized pathogenic variant. Eur J Clin Microbiol Infect Dis 31:981–989. https://doi.org/10.1007/s10096-011-1396-6
Shon AS, Bajwa RPS, Russo TA (2013) Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: A new and dangerous breed. Virulence 4:107–118. https://doi.org/10.4161/viru.22718
Turton JF, Englender H, Gabriel SN, Turton SE, Kaufmann ME, Pitt TL (2007) Genetically similar isolates of Klebsiella pneumoniae serotype K1 causing liver abscesses in three continents. J Med Microbiol 56:593–597. https://doi.org/10.1099/jmm.0.46964-0
Melzer M, Petersen I (2007) Mortality following bacteraemic infection caused by extended spectrum beta-lactamase (ESBL) producing E. coli compared to non-ESBL producing E. coli. J Infect 55:254–259. https://doi.org/10.1016/j.jinf.2007.04.007
Price DB, Honeybourne D, Little P, Mayon-White RT, Read RC, Thomas M et al (2004) Community-acquired pneumonia mortality: a potential link to antibiotic prescribing trends in general practice. Respir Med 98:17–24. https://doi.org/10.1016/j.rmed.2003.08.011
Blomberg B, Manji KP, Urassa WK, Tamim BS, Mwakagile DSM, Jureen R et al (2007) Antimicrobial resistance predicts death in Tanzanian children with bloodstream infections : a prospective cohort study. BMC Infect Dis 7:1–14. https://doi.org/10.1186/1471-2334-7-43
Marra AR, Pereira CAP, Castelo A, Do Carmo Filho JR, Cal RGR, Sader HS et al (2006) Health and economic outcomes of the detection of Klebsiella pneumoniae-produced extended-spectrum β-lactamase (ESBL) in a hospital with high prevalence of this infection. Int J Infect Dis 10:56–60. https://doi.org/10.1016/j.ijid.2005.04.002
Hu B, Ye H, Xu Y, Ni Y, Hu Y, Yu Y et al (2010) Clinical and economic outcomes associated with community-acquired intra-abdominal infections caused by extended spectrum beta-lactamase (ESBL) producing bacteria in China. Curr Med Res Opin 26:1443–1449. https://doi.org/10.1185/03007991003769068
Esteve-Palau E, Solande G, Sánchez F, Sorlí L, Montero M, Güerri R et al (2015) Clinical and economic impact of urinary tract infections caused by ESBL-producing Escherichia coli requiring hospitalization: a matched cohort study. J Infect 71:667–674. https://doi.org/10.1016/j.jinf.2015.08.012
Pitout JDD, Nordmann P, Poirel L (2015) Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob Agents Chemother 59:5873–5884. https://doi.org/10.1128/AAC.01019-15
Yeo JM (2016) Antimicrobial stewardship: improving antibiotic prescribing practice in a respiratory ward. BMJ Qual Improv Rep 5:u206491.w3570. https://doi.org/10.1136/bmjquality.u206491.w3570
Bantar C, Sartori B, Vesco E, Heft C, Saúl M, Salamone F et al (2003) A hospitalwide intervention program to optimize the quality of antibiotic use: impact on prescribing practice, antibiotic consumption, cost savings, and bacterial resistance. Clin Infect Dis 37:180–186. https://doi.org/10.1086/375818
Hardy-Holbrook R, Aristidi S, Chandnani V, Dewindt D, Dinh K (2013) Antibiotic resistance and prescribing in Australia: current attitudes and practice of GPs. Healthc Infect 18:147–151. https://doi.org/10.1071/HI13019
Gwimile JJ, Shekalaghe SA, Kapanda GN, Kisanga ER (2012) Antibiotic prescribing practice in management of cough and/or diarrhoea in Moshi municipality, Northern Tanzania: cross-sectional descriptive study. Pan Afr Med J 12:103
Thriemer K, Katuala Y, Batoko B, Alworonga J-P, Devlieger H, Van Geet C et al (2013) Antibiotic prescribing in DR Congo: a knowledge, attitude and practice survey among medical doctors and students. PLoS One 8:e55495. https://doi.org/10.1371/journal.pone.0055495
Ah YM, Kim AJ, Lee JY (2014) Colistin resistance in Klebsiella pneumoniae. Int J Antimicrob Agents 44:8–15. https://doi.org/10.1016/j.ijantimicag.2014.02.016
van den Boogaard J, Semvua HH, Boeree MJ, Aarnoutse RE, Kibiki GS (2009) Sale of fluoroquinolones in northern Tanzania: a potential threat for fluoroquinolone use in tuberculosis treatment. J Antimicrob Chemother 65:145–147. https://doi.org/10.1093/jac/dkp413
Sonda T, Kumburu H, van Zwetselaar M, Ahrenfeldt J, Alifrangis M, Lund O et al (2016) Benchtop whole-genome sequencing for identification of nosocomial outbreaks in Tanzania. Infect Control Hosp Epidemiol 37:622–623. https://doi.org/10.1017/ice.2016.28
van Zwetselaar M, Nyombi B, Sonda T, Kumburu H, Chamba N, Dekker MCJ et al (2018) Aeromonas caviae mimicking Vibrio cholerae infectious enteropathy in a cholera-endemic region with possible public health consequences: two case reports. J Med Case Rep 12:71. https://doi.org/10.1186/s13256-018-1603-5
Kumburu HH, Sonda T, Mmbaga BT, Alifrangis M, Lund O, Kibiki G et al (2017) Patterns of infections, aetiological agents and antimicrobial resistance at a tertiary care hospital in northern Tanzania. Tropical Med Int Health 22:454–464. https://doi.org/10.1111/tmi.12836
Illumina®. Nextera® XT library prep reference guide [Internet]. Document #15031942 v01 2016;1–28. doi: http://support.illumina.com/downloads/nextera_xt_sample_preparation_guide_15031942.html
Andrews S, FastQC A (2018) Quality control tool for high throughput sequence data
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. https://doi.org/10.1093/bioinformatics/btt086
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O et al (2012) Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. https://doi.org/10.1093/jac/dks261
Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL et al (2012) Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361. https://doi.org/10.1128/JCM.06094-11
Carattoli A, Zankari E, Garciá-Fernández A, Larsen MV, Lund O, Villa L et al (2014) In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903. https://doi.org/10.1128/AAC.02412-14
Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM et al (2014) Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol 52:1501–1510. https://doi.org/10.1128/JCM.03617-13
Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O (2014) Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. https://doi.org/10.1371/journal.pone.0104984
Souverein D, Boers SA, Veenendaal D, Euser SM, Kluytmans J, Den Boer JW (2014) Polyclonal spread and outbreaks with ESBL positive gentamicin resistant Klebsiella spp. in the region Kennemerland, the Netherlands. PLoS One 9:e101212. https://doi.org/10.1371/journal.pone.0101212
Löhr IH, Hülter N, Bernhoff E, Johnsen PJ, Sundsfjord A, Naseer U (2015) Persistence of a pKPN3-like CTX-M-15-encoding IncFIIK plasmid in a Klebsiella pneumonia ST17 host during two years of intestinal colonization. PLoS One 10:e0116516. https://doi.org/10.1371/journal.pone.0116516
Zhou K, Lokate M, Deurenberg RH, Tepper M, Arends JP, Raangs EGC et al (2016) Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae. Sci Rep 6:20840. https://doi.org/10.1038/srep20840
Snitkin ES, Zelazny AM, Thomas PJ, Stock F, Henderson DK, Palmore TN et al (2012) Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116–148ra116. https://doi.org/10.1126/scitranslmed.3004129
Becker L, Fuchs S, Pfeifer Y, Semmler T, Eckmanns T, Korr G et al (2018) Whole genome sequence analysis of CTX-M-15 producing klebsiella isolates allowed dissecting a polyclonal outbreak scenario. Front Microbiol 9:322. https://doi.org/10.3389/fmicb.2018.00322
Taitt CR, Leski TA, Erwin DP, Odundo EA, Kipkemoi NC, Ndonye JN et al (2017) Antimicrobial resistance of Klebsiella pneumoniae stool isolates circulating in Kenya. PLoS One 12:e0178880. https://doi.org/10.1371/journal.pone.0178880
Tellevik MG, Blomberg B, Kommedal Ø, Maselle SY, Langeland N, Moyo SJ (2016) High prevalence of faecal carriage of esbl-producing enterobacteriaceae among children in Dar Es Salaam, Tanzania. PLoS ONE 11:e0168024. https://doi.org/10.1371/journal.pone.0168024
Sonda T, Kumburu H, van Zwetselaar M, Alifrangis M, Mmbaga BT, Aarestrup FM et al (2018) Whole genome sequencing reveals high clonal diversity of Escherichia coli isolated from patients in a tertiary care hospital in Moshi, Tanzania. Antimicrob Resist Infect Control 7:72. https://doi.org/10.1186/s13756-018-0361-x
Di Mento G, Cuscino N, Carcione C, Cardinale F, Conaldi PG, Douradinha B (2017) Emergence of a Klebsiella pneumoniae ST392 clone harbouring KPC-3 in an Italian transplantation hospital. J Hosp Infect 98:313–314. https://doi.org/10.1016/j.jhin.2017.11.019
Vubil D, Figueiredo R, Reis T, Canha C, Boaventura L, Da Silva GJ (2017) Outbreak of KPC-3-producing ST15 and ST348 Klebsiella pneumoniae in a Portuguese hospital. Epidemiol Infect 145:595–599. https://doi.org/10.1017/S0950268816002442
Coelho A, González-López JJ, Miró E, Alonso-Tarrés C, Mirelis B, Larrosa MN et al (2010) Characterisation of the CTX-M-15-encoding gene in Klebsiella pneumoniae strains from the Barcelona metropolitan area: plasmid diversity and chromosomal integration. Int J Antimicrob Agents 36:73–78. https://doi.org/10.1016/j.ijantimicag.2010.03.005
Madoshi BP, Kudirkiene E, Mtambo MMA, Muhairwa AP, Lupindu AM, Olsen JE (2016) Characterisation of commensal Escherichia coli isolated from apparently healthy cattle and their attendants in Tanzania. PLoS ONE 11:e0168160. https://doi.org/10.1371/journal.pone.0168160
Holt KE, Wertheim H, Zadoks RN, Baker S, Whitehouse CA, Dance D et al (2015) Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci U S A 112:E3574–E3581. https://doi.org/10.1073/pnas.1501049112
Li J, Ren J, Wang W, Wang G, Gu G, Wu X et al (2017) Risk factors and clinical outcomes of hypervirulent Klebsiella pneumoniae induced bloodstream infections. Eur J Clin Microbiol Infect Dis 37:679–689. https://doi.org/10.1007/s10096-017-3160-z
Bialek-Davenet S, Criscuolo A, Ailloud F, Passet V, Jones L, Delannoy-Vieillard AS et al (2014) Genomic definition of hypervirulent and multidrug-resistant klebsiella pneumoniae clonal groups. Emerg Infect Dis 20:1812–1820. https://doi.org/10.3201/eid2011.140206
Ballén V, Sáez E, Benmessaoud R, Houssain T, Alami H, Barkat A et al (2015) First report of a Klebsiella pneumoniae ST466 strain causing neonatal sepsis harbouring the blaCTX-M-15gene in Rabat, Morocco. FEMS Microbiol Lett 362:1–4. https://doi.org/10.1093/femsle/fnu026
Acknowledgements
We thank the management of Kilimanjaro Christian Medical Centre and all patients who consented to participate in this study.
Funding
This study was supported by DANIDA through Danida Fellowship Centre award number DFC no. 12-007DTU.
Author information
Authors and Affiliations
Contributions
TS conceived the initial idea; FA, OL, MA, BTM and GK refined the idea. TS and HK performed the laboratory analyses. TS and MZ analysed the data and prepared the manuscript draft. TS, HK, MZ, FA, OL, MA, BTM and GK read, revised and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and participant’s consent
This study was granted ethical approval by the KCMUCo Research Ethics Committee and the National Institute for Medical Research with approval numbers 893 and NIMR/HQ/R.8a/Vol.IX/2080, respectively. A written informed consent was obtained from each participant or from parents or guardians of children before enrolment into the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Availability of Data and Materials
Genome assemblies have been submitted to the European Nucleotide Archive with project accession number PRJEB26616. Other data are available on request to the authors.
Electronic supplementary material
ESM 1
(XLS 35 kb)
Rights and permissions
About this article

Cite this article
Sonda, T., Kumburu, H., van Zwetselaar, M. et al. Molecular epidemiology of virulence and antimicrobial resistance determinants in Klebsiella pneumoniae from hospitalised patients in Kilimanjaro, Tanzania. Eur J Clin Microbiol Infect Dis 37, 1901–1914 (2018). https://doi.org/10.1007/s10096-018-3324-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-018-3324-5