
Abstract
High-resolution screening methodologies which enable the differentiation of Chlamydia trachomatis at the strain level, directly from clinical samples, can provide the detailed information required for epidemiological questions such as the dynamics of treatment failure. In addition, they give a detailed snapshot of circulating C. trachomatis genetic variation, data which are currently lacking for the Australian population. In the context of two Australian clinical trials, we assessed the genetic diversity of C. trachomatis and compared these to strains circulating globally. We used high-resolution multilocus sequence typing (MLST) of five highly variable genetic regions of C. trachomatis to examine variation in Australia. Samples with established genovars were drawn from a pool of 880 C. trachomatis-positive samples from two clinical studies, whereby 76 sample pairs which remained C. trachomatis-positive for the same genovar after treatment underwent MLST analysis to distinguish between treatment failure and reinfection. MLST analysis revealed a total of 25 sequence types (STs), six new allele variants and seven new STs not described anywhere else in the world, when compared to those in the international C. trachomatis MLST database. Of the eight most common global STs, seven were found in Australia (four derived from men who have sex with men (MSM) and three from heterosexuals). Newly identified STs were predominantly found in samples from the MSM population. In conclusion, MLST provided a diverse C. trachomatis strain profile, with novel circulating STs, and could be used to identify local sexual networks to focus on interventions such as testing and partner notification to prevent reinfection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chlamydia trachomatis is one of the most prevalent bacterial sexually transmitted infections (STIs) worldwide [1]. C. trachomatis genovars A–C are primarily associated with trachoma, genovars D–K with sexually transmitted urogenital disease [2] and genovars L1–L3 are responsible for a painful and often serious condition known as lymphogranuloma venereum (LGV) [3]. Within Australia, the prevalence of anogenital chlamydia is greatest among young adults (<25 years old), men who have sex with men (MSM) and indigenous people who also have endemic rates of ocular C. trachomatis in remote regions of the Northern Territory [4, 5].
The diagnosis of C. trachomatis is now predominantly based on molecular detection of the organism alone. Generally, the organism is not propagated, so traditional serotyping based on the antigenic properties of the major outer membrane protein (MOMP) is no longer possible. Instead, serovar prediction is now based on distinct nucleotide sequence variations within the ompA gene (which encodes MOMP) [6]. The application of specific primer and probe combinations [7] enables the detection of what are referred to as genovars. Genovars provide additional information which facilitate monitoring of sexual networks, incidence of treatment failure and reinfection, allowing refinement of treatment recommendations. Recent studies show that the three most dominant genovars of C. trachomatis are D, E and F, with E and F most frequently detected in the heterosexual population worldwide, including Australia [6, 8, 9] (E 49%, F 20%, D 8.1%, G 9.8% [9, 10]) and genovars D, G and J being the dominant types in MSM [8, 11] worldwide, including Australia [12]. The prevalence of these genovars has stayed relatively constant within the Australian MSM population, currently identified at a prevalence of D (29%), G (35%) and J (16–20%) [9, 13].
Although ompA genovar typing can be useful in a diagnostic setting, lack of genetic variation within individual genovars limits the discriminatory power to identify variation between strains. In addition, whole genome analysis of C. trachomatis using high-concentration culture-derived samples indicates that recombination events make ompA alone a poor candidate for genetic strain typing [14]. Resolution at the strain level is required to accurately determine the geographical distribution within and between genovars, transmission patterns within sexual networks or to enable the distinction between types of treatment failure; persistent, new infection or reinfection within the clinical setting. Although whole genome sequencing is the pinnacle for genetic resolution analysis, this requires either the capture or culture of this organism from clinical samples to achieve satisfactory yields of C. trachomatis-specific sample, making this approach not feasible for most laboratories. Furthermore, due to the limitations associated with ompA genovar typing, high resolution multilocus sequence typing (MLST) of C. trachomatis provides a cost-effective screening approach that can be applied to a range of epidemiological and clinical applications beyond ompA genovar typing [6, 15–17]. The resolution obtained by sequencing the five highly variable MLST regions of C. trachomatis was recently reported to be 2.1 times higher than ompA sequence determination alone [18]. It has also been indicated that phylogenetic trees obtained from MLST data were similar to those obtained from whole genome analysis [8].
In the context of two Australian clinical studies, we conducted an MLST analysis of C. trachomatis-positive samples to evaluate chlamydial genetic diversity circulating predominantly within the MSM population, and a small heterosexual population, and compare these to strains circulating globally.
Methods
Sample population
The samples included in this study were taken from two previously published studies [13, 19]. The REACT study was a randomised controlled trial of SMS reminders and home collection kits to retest individuals treated for chlamydia, conducted from 2011 to 2014 in sexual health services in Melbourne and Sydney [19]. The REARS study used stored chlamydia-positive rectal samples from MSM collected at Melbourne Sexual Health Centre between July 2008 and October 2013. In both studies, MLST analysis was undertaken on individuals who had two positive samples with the same genovar separated by 120 (REACT) or 100 (REARS) days to determine the cause of the second positive sample (e.g. new infection, reinfection or treatment failure). These results are reported elsewhere [9, 10, 13].
Inclusion of MLST results in this current study, for sequence type (ST) analysis only, were based on the following criteria: (i) MLST STs were complete for all five regions, (ii) where an individual has identical STs for both samples, only one ST was included in the analysis, (iii) where an individual has different STs for both samples, both STs were included in the analysis.
The REACT and REARS studies received ethical approval from the Alfred Health Human Research Ethics Committee, South Eastern Sydney and Illawarra Area Health Service Human Research Ethics Committee and University of New South Wales Human Research Ethics Committee.
PCR amplification, genovar and MLST
Nucleic acid was isolated from a 200 μL aliquot of each of the 49 REARS sample pairs (n = 98) utilising the MagNA Pure 96 system (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s instructions and subsequently eluted in 100 μL of MagNA Pure 96 elution buffer, as described previously [13]. The 27 REACT sample pairs (n = 54) were extracted using the MagNA Pure 96 system as described above, with the exception of those samples in Cobas media. For these, the Roche Cobas 4800 platform (Roche Molecular Diagnostics, Branchburg, NJ 08876, USA) removed 400 μL of sample (vaginal or rectal swabs) or 850 μL of sample (urine) for the extraction of nucleic acid, which is subsequently eluted into a final volume of 100 μL of elution buffer.
Quantitative real-time polymerase chain reaction (PCR) was performed as described [7] to classify positive samples into one of three broad phylogenetic groups. A secondary set of PCR assays was then used to determine specific genovars [7] for each sample. Subsequently, the 76 pre- and post-treatment samples (n = 152) with the same genovar were amplified for five of the most variable regions of the C. trachomatis genome: hctB, CT058, CT144, CT172 and pbpB, according to the current protocol provided at http://mlstdb.bmc.uu.se [6]. PCR amplicons for each region were quantified and then sequenced in both directions using Sanger sequencing (Australian Genome Research Facility, Melbourne, Australia). The reference strain D/UW-3/CX (GenBank: AE001273) was used as a control throughout.
Sequence analysis
Sequence analysis was performed using the CLC Main Workbench (version 7, CLC Bio, Aarhus, Denmark). The Uppsala University C. trachomatis MLST database (http://mlstdb.bmc.uu.se) was utilised to perform allele sequence and ST queries to identify and compare allele and ST profiles to those previously identified. Alleles which were not an identical match to those in the database were aligned with alleles of the closest match using either CLC Main Workbench or MEGA6 [20], to determine variant sites. Unique sequence variations were submitted to the Uppsala University C. trachomatis MLST database for confirmation and denoted unique allele and ST numbers.
MST, eBURST and phylogenetic analysis
MLST data were analysed using BioNumerics 7.5 (Applied Maths, Sint-Martens-Latem, Belgium) to create a minimum spanning tree (MST), which was partitioned into clusters. A cluster was defined as C. trachomatis STs that differ by a single locus variation, whereas partitioning of clusters was defined by variation at two or more loci between STs.
eBURST version 3 software (http://eburst.mlst.net/) was used to identify founder STs within the Australian dataset and define groups and singletons. Analysis was performed on the eBURST website using the single dataset function whereby STs for all five alleles were specified and the number of loci set to 5. The analysis generates groups with predicted founders and lists singleton STs, with a singleton defined as an ST with no single locus variants.
Phylogenetic analysis was based on concatenated allele sequences from each of the five regions, for each ST identified in this study and eight reference strains representing genovars. The STs for the reference strains and the previously known samples were entered into the http://pubmlst.org/ database. Using the MLST (C. trachomatis Uppsala) scheme, selecting all loci and the concatenate in frame option, we extracted the concatenated sequences as FASTA files and uploaded them into the MEGA7 software [21]. As the Australian STs identified in this study are not yet in the http://pubmlst.org/ database, these allele sequences were concatenated manually (in the same order as the pubMLST database) and aligned using MEGA7. A phylogenetic tree for 33 concatenated STs was constructed with MEGA7, whereby evolutionary history was inferred by using the maximum likelihood method based on the Tamura–Nei model [22]. Support values were estimated using 1000 bootstraps and the tree was rooted using the LGV strains as the outgroup. Reference genomes were chosen based on their presence within the Uppsala MLST database.
Results
From the 76 samples, 82% (n = 62) were from Victoria and 18% (n = 14) were from NSW. Samples were primarily MSM (n = 64; 84%) and 12 (16%) heterosexuals (six female and six male). The proportion of C. trachomatis ompA genovars by risk group for MSM were G (32%), D (25%) and J (15%), with E, B, L2 and F also present. Among heterosexuals, 75% were E and 25% were F.
Of the 76 sample pairs for which MLST analysis was performed, five sample pairs had STs which were not complete for all five regions sequenced, and so, were excluded based on criteria (i). Of the 71 sample pairs with complete STs, 66 sample pairs had identical STs, 25 from REACT [9, 10] and 41 from REARS [13]. According to criteria (ii), to avoid duplicate sampling of STs for each individual, only one ST from each of these 66 sample pairs was included in the analysis. Finally, five individuals had different STs for the sample pair, so in accordance to criteria (iii), both STs (n = 10) were included in the analysis. This gave a total of 76 STs [66 from criteria (ii) and ten from criteria (iii)] that were used for MST analysis.
Using the Uppsala University C. trachomatis MLST database to query alleles and STs, new variants from those previously reported were identified. Alleles which were not an identical match to those in the C. trachomatis MLST database were aligned with alleles of the closest match using either CLC Main Workbench or MEGA6 to determine variant sites. Six allele variants were identified within these Australian samples which had not been previously described. At least one allele variation was identified in each of the five MLST regions sequenced, except CT172. The position of each variant given was derived from alignment with the allele sequence which had the highest percentage similarity in the C. trachomatis MLST database for that region (Table 1). Allele variations were assigned nomenclature as denoted in Table 1.
A total of 25 different STs were identified in this study. Of these, 18 STs have been described previously (http:/mlstdb.bmc.uu.se) and seven were new STs. Of these seven new STs, five were identified within MSM with three predominating, ST570 (n = 5) and ST564 and ST569 (each n = 4), while ST565 (n = 2) and ST567 (n = 1) were less common. Two new STs were identified within the heterosexual group ST566 and ST568 (each n = 1). Of the STs reported previously, ST109 (n = 12), ST33 (n = 8) and ST108 (n = 5) were found in samples from MSM only. Within the small heterosexual subset, ST3 (n = 3), ST12 and ST59 (each n = 2) were the most common. ST3 and ST56 were observed in both MSM and heterosexuals.
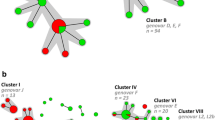
In total, 76 STs were used to generate an MST. The distribution of these STs is illustrated in Fig. 1. The MST shows two main branches; one with genovars E, F and D which contains all samples from heterosexuals and the other is comprised of genovars G, J, B and L2 being derived from MSM samples only, with the connecting genovar D also from MSM only. The MST is further divided onto clusters which represent genetically related C. trachomatis strain types whose STs differ at only one allele region. Cluster I was the largest, comprised of ten STs and 32 samples. Clusters II, V, VI and VII are all represented by a single ST, with cluster VII being the most distant, differing at all five allele regions from the other STs. The new STs identified within this Australian population are represented randomly throughout the MST. The distribution of ST clusters was reflected in the phylogenetic tree shown in Fig. 2. The LGV strains are again the most distant, representing a separate clade. The other STs show three subclades: (a) two reference STs (E_ST55, F_ST12) grouped with clusters III and IV, respectively, with D_ST20, G_ST278 on a separate branch, (b) reflects MSM only clusters with J_ST136, (c) C_ST46 represents the only ocular strain ST as distant to subclade b.

Minimum spanning tree (MST) of 76 Chlamydia trachomatis sequence types (STs) from Australia. Each circle represents a multilocus sequence typing (MLST) ST. The size of the circle is directly proportional to the number of samples with an identical ST. The number shown within each circle denotes the ST. White circles indicate new STs identified in Australia and blue circles indicate previously identified STs. Bold lines between circles indicate STs that differ by a single locus variation. Thin lines indicate STs which have 2–4 variant loci and the transparent dotted line indicates variation at all five loci. Grey shading indicates clusters

Chlamydia trachomatis ST phylogeny. Maximum likelihood inferred phylogeny of 33 concatenated sequences for the five MLST allele regions. The percentage of trees in which the associated STs clustered together is shown next to the branches and branch lengths are measured by the number of substitutions per site. The tree was rooted by the L strain ST branch. The urogenital strains are divided into two subclades based on ST. (a) pink represents STs identified in the mixed (heterosexual/MSM) cluster, (b) green represents STs associated with MSM only. The only ocular strain ST is shown in yellow (c)
Analysis of the 25 STs using eBURST predicted two founding groups, group 1 ST108 with ST52 as a subgroup (both grouping with subclade b) and group 2 ST56 (groups with subclade a). The two founder groups are at opposite ends of the MST, with ST108 and ST52 forming the central part of the largest MSM only cluster (I). Comparatively, the predicted founder of group 2, ST56 formed cluster III, comprised of both MSM and heterosexuals. Although ST109 was the most common ST, being one of six singletons, it was not identified as a founder by eBURST.
Discussion
To our knowledge, this is the first comprehensive study reporting the circulating genetic variation in C. trachomatis at the strain level within Australia. The results presented indicate that Australian ST profiles reflect the global distribution of genovars and founder STs for both MSM and heterosexuals [8, 18], in addition to other predominant STs (ST33 in MSM, ST59 in heterosexuals). The existence of localised genetic diversity of C. trachomatis strains circulating within Australia demonstrates local divergence, with new alleles and STs identified.
Previous studies have used MLST to estimate the geographical distribution, transmission and prevalence of C. trachomatis strains [15, 17, 23], the most recent of which provided an overview of C. trachomatis strains circulating globally from 16 countries [8]. Interestingly, the distribution of MLST STs demonstrates that there are eight common STs which are distributed globally, four of which were strongly associated with MSM (ST52, ST58, ST108, ST109) and four associated with heterosexuals (ST3, ST12, ST55, ST56). These common STs are reflected in Australia, with the exception of ST55 in the heterosexual group, the most likely reason for this being the small number of heterosexuals within this sample set. However, prevalence data on C. trachomatis strains in the southern hemisphere are lacking, with only a small number of samples from Argentina and a handful from Chile and Australia [8].
Our study and another recent study [18] are beginning to expand the southern hemisphere dataset. As described by Isaksson et al. [18] in Argentina and Chile, the dominant ompA genovar was E, with the most common STs (ST3 and ST56) identified as founders [18]. These STs were also the most frequent in the Australian population (ST56 32%; ST3 25%) for genovar E (with two new STs and seven STs overall), with ST56 identified as a founder in the heterosexual/MSM group. Australian STs for the other two predominant genovars G and D had no overlap with those described by Isaksson et al. [18]. The majority of individuals with genovar D had ST109 common to MSM (80%), with ST11 being the only other in this group. The most prevalent genovar in the Australian dataset was G, which had the most diversity with nine STs, the most common being ST33 (38%), followed by the new ST569 (19%).
The 25 Australian STs identified in this study formed seven clusters (Fig. 1). The distribution of these clusters is similar to those observed previously [8], whereby STs on one end of the MST represent those commonly observed in MSM and those on the other end represent those often observed in heterosexuals (although here, both MSM and heterosexuals are represented). Interestingly, ST109 forms the bridging cluster (Australian dataset: cluster II, genovar D, ST109) between the MSM STs and heterosexual STs, which is also evident in the two recent MLST studies incorporating global data [8, 18]. Although ST109 is a singleton and made up of MSM, it appears to be the main divergence point between the two sexual groups in each of these datasets. Further, eBURST analysis identified two founding groups, group 1 with one subgroup at the MSM only side of the MST and group 2 at the other end with both heterosexual and MSM STs. Phylogenetic analysis of the concatenated sequences for these STs reflects the distribution of the MST clusters, further illustrating the novel lineages in this dataset (Fig. 2). Interestingly, four of the reference STs group with the mixed sexual group, with only one reference ST grouping with MSM and the one ocular reference ST included forming a subgroup off this. Comparison with phylogenetic trees from previous studies is limited here; as the genovar-based ompA sequence was not generated for this study, we were unable to include these in our phylogenetic analysis.
There were other limitations of this study. First, as a result of the studies from which the samples were sourced, this study is biased towards MSM and under-represents heterosexuals. Although the distribution of ompA genovars reflects those commonly found to be associated with heterosexuals, the diversity of STs within this group is likely under-represented. Second, our samples are not a random selection of chlamydia-positive samples but, rather, samples were selected for MLST on the basis of repeated detection of an identical genovar (same strain for treatment failure or partner reinfection, different strain if new infection). If treatment failure is related to chlamydia type, as evidenced by MLST, then this study will be biased towards such samples. Third, we used clinical samples, which means that some of those selected for MLST did not deliver a full complement of STs, and, so, could not be included.
Genital chlamydia infection became a notifiable disease in 1991 in Australia [24]. The use of discriminatory genotyping tools (MLST) which can differentiate at the strain level provides valuable epidemiological information about local populations and the dynamics of C. trachomatis spread within these, in addition to identifying the most common circulating types within sexual networks. Further, MLST can be a valuable tool to help differentiate between reinfection and persistent infection or treatment failure, particularly as there is growing concern about azithromycin treatment failure for urogenital and anogenital chlamydia infection.
In conclusion, although the genovar distribution of the Australian population is similar to that globally, the MLST strain types suggest that distributions of non-founder STs have both known and new STs, creating distinct local population signatures. This study provides a snapshot of several eastern Australian regions. Given the lack of data pertaining to C. trachomatis strain distribution within this region, further comprehensive studies within the western Pacific region are warranted. Based on the discriminatory power of MLST data, further larger studies using this methodology would be useful. The need for more rapid and affordable screening methods directly from clinical samples remains, particularly if emerging antibiotic resistance is on the rise.
References
World Health Organization (WHO) (2012) Global incidence and prevalence of selected curable sexually transmitted infections—2008. WHO, Geneva. Available online at: http://www.who.int/reproductivehealth/publications/rtis/2008_STI_estimates.pdf
Garland SM, Malatt A, Tabrizi S, Grando D, Lees MI, Andrew JH, Taylor HR (1995) Chlamydia trachomatis conjunctivitis. Prevalence and association with genital tract infection. Med J Aust 162(7):363–366
Geisler WM, Suchland RJ, Whittington WL, Stamm WE (2003) The relationship of serovar to clinical manifestations of urogenital Chlamydia trachomatis infection. Sex Transm Dis 30(2):160–165
Andersson P, Harris SR, Seth Smith HM, Hadfield J, O’Neill C, Cutcliffe LT, Douglas FP, Asche LV, Mathews JD, Hutton SI, Sarovich DS, Tong SY, Clarke IN, Thomson NR, Giffard PM (2016) Chlamydia trachomatis from Australian Aboriginal people with trachoma are polyphyletic composed of multiple distinctive lineages. Nat Commun 7:10688. doi:10.1038/ncomms10688
Lewis D, Newton DC, Guy RJ, Ali H, Chen MY, Fairley CK, Hocking JS (2012) The prevalence of Chlamydia trachomatis infection in Australia: a systematic review and meta-analysis. BMC Infect Dis 12:113. doi:10.1186/1471-2334-12-113
Bom RJ, Christerson L, Schim van der Loeff MF, Coutinho RA, Herrmann B, Bruisten SM (2011) Evaluation of high-resolution typing methods for Chlamydia trachomatis in samples from heterosexual couples. J Clin Microbiol 49(8):2844–2853. doi:10.1128/JCM.00128-11
Stevens MP, Twin J, Fairley CK, Donovan B, Tan SE, Yu J, Garland SM, Tabrizi SN (2010) Development and evaluation of an ompA quantitative real-time PCR assay for Chlamydia trachomatis serovar determination. J Clin Microbiol 48(6):2060–2065. doi:10.1128/JCM.02308-09
Herrmann B, Isaksson J, Ryberg M, Tångrot J, Saleh I, Versteeg B, Gravningen K, Bruisten S (2015) Global multilocus sequence type analysis of Chlamydia trachomatis strains from 16 countries. J Clin Microbiol 53(7):2172–2179. doi:10.1128/JCM.00249-15
Smith KS, Guy R, Danielewski JA, Tabrizi SN, Chen M, Kaldor JM, Hocking JS (2015) High chlamydia treatment failure rates in men who have sex with men. In: Proceedings of the World STI & HIV Congress, Brisbane, Australia, September 2015, p O15.13
Smith KS GR, Danielewski JA, Tabrizi SN, Fairley CK, McNulty AM, Rawlinson W, Saville M, Garland SM, Donovan B, Kaldor JM, Hocking JS (2017) Biological and behavioural factors associated with positive chlamydia retests. Sex Transm Dis (in press)
Bom RJ, Matser A, Bruisten SM, van Rooijen MS, Heijman T, Morré SA, de Vries HJ, Schim van der Loeff MF (2013) Multilocus sequence typing of Chlamydia trachomatis among men who have sex with men reveals cocirculating strains not associated with specific subpopulations. J Infect Dis 208(6):969–977. doi:10.1093/infdis/jit275
Twin J, Moore EE, Garland SM, Stevens MP, Fairley CK, Donovan B, Rawlinson W, Tabrizi SN (2011) Chlamydia trachomatis genotypes among men who have sex with men in Australia. Sex Transm Dis 38(4):279–285. doi:10.1097/OLQ.0b013e3181fc6944
Kong FY, Tabrizi SN, Fairley CK, Phillips S, Fehler G, Law M, Vodstrcil LA, Chen M, Bradshaw CS, Hocking JS (2016) Higher organism load associated with failure of azithromycin to treat rectal chlamydia. Epidemiol Infect 144(12):2587–2596. doi:10.1017/S0950268816000996
Harris SR, Clarke IN, Seth-Smith HM, Solomon AW, Cutcliffe LT, Marsh P, Skilton RJ, Holland MJ, Mabey D, Peeling RW, Lewis DA, Spratt BG, Unemo M, Persson K, Bjartling C, Brunham R, de Vries HJ, Morré SA, Speksnijder A, Bébéar CM, Clerc M, de Barbeyrac B, Parkhill J, Thomson NR (2012) Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet 44(4):413–419, S1. doi:10.1038/ng.2214
Gravningen K, Christerson L, Furberg AS, Simonsen GS, Ödman K, Ståhlsten A, Herrmann B (2012) Multilocus sequence typing of genital Chlamydia trachomatis in Norway reveals multiple new sequence types and a large genetic diversity. PLoS One 7(3), e34452. doi:10.1371/journal.pone.0034452
Klint M, Fuxelius HH, Goldkuhl RR, Skarin H, Rutemark C, Andersson SG, Persson K, Herrmann B (2007) High-resolution genotyping of Chlamydia trachomatis strains by multilocus sequence analysis. J Clin Microbiol 45(5):1410–1414. doi:10.1128/JCM.02301-06
Versteeg B, Himschoot M, van den Broek IV, Bom RJ, Speksnijder AG, Schim van der Loeff MF, Bruisten SM (2015) Urogenital Chlamydia trachomatis strain types, defined by high-resolution multilocus sequence typing, in relation to ethnicity and urogenital symptoms among a young screening population in Amsterdam, The Netherlands. Sex Transm Infect 91(6):415–422. doi:10.1136/sextrans-2014-051790
Isaksson J, Gallo Vaulet L, Christerson L, Ruettger A, Sachse K, Entrocassi C, Castro É, Rodríguez Fermepin M, Herrmann B (2016) Comparison of multilocus sequence typing and multilocus typing microarray of Chlamydia trachomatis strains from Argentina and Chile. J Microbiol Methods 127:214–218. doi:10.1016/j.mimet.2016.06.005
Smith KS, Hocking JS, Chen M, Fairley CK, McNulty A, Read P, Bradshaw CS, Tabrizi SN, Wand H, Saville M, Rawlinson W, Garland SM, Donovan B, Kaldor JM, Guy R (2014) Rationale and design of REACT: a randomised controlled trial assessing the effectiveness of home-collection to increase chlamydia retesting and detect repeat positive tests. BMC Infect Dis 14:223. doi:10.1186/1471-2334-14-223
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729. doi:10.1093/molbev/mst197
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. doi:10.1093/molbev/msw054
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10(3):512–526
Bom RJ, van der Helm JJ, Bruisten SM, Grünberg AW, Sabajo LO, Schim van der Loeff MF, de Vries HJ (2013) The role of Surinamese migrants in the transmission of Chlamydia trachomatis between Paramaribo, Suriname and Amsterdam, The Netherlands. PLoS One 8(11), e77977. doi:10.1371/journal.pone.0077977
NNDSS Annual Report Writing Group, Newman L, Stirzaker S, Knuckey D, Robinson K, Hood J, Knope K, Fitzsimmons G, Barker S, Martin N, Siripol S, Gajanayake I, Kaczmarek M, Barr I, Hii A, Foxwell R, Owen R, Liu C, Wright P, Sanders L, Barry C, Ormond J (2010) Australia’s notifiable disease status, 2008: annual report of the National Notifiable Diseases Surveillance System. Commun Dis Intell Q Rep 34(3):157–224
Acknowledgements
We would like to thank Jenny Isaksson from Uppsala University, Sweden for her assistance with assigning new MLST alleles and STs nomenclature in the Uppsala University C. trachomatis MLST database (http://mlstdb.bmc.uu.se).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by an Australian National Health and Medical Research Council program grant no. 568971.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Ethics approval for the REACT and REARS studies was received from the Alfred Health Human Research Ethics Committee, South Eastern Sydney and Illawarra Area Health Service Human Research Ethics Committee and University of New South Wales Human Research Ethics Committee.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Danielewski, J.A., Phillips, S., Kong, F.Y.S. et al. A snapshot of Chlamydia trachomatis genetic diversity using multilocus sequence type analysis in an Australian metropolitan setting. Eur J Clin Microbiol Infect Dis 36, 1297–1303 (2017). https://doi.org/10.1007/s10096-017-2935-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-017-2935-6